





Abstract
The fifth edition of the WHO Classification of Tumors of the Central Nervous System (CNS), published in 2021, is the sixth version of the international standard for the classification of brain and spinal cord tumors. Building on the 2016 updated fourth edition and the work of the Consortium to Inform Molecular and Practical Approaches to CNS Tumor Taxonomy, the 2021 fifth edition introduces major changes that advance the role of molecular diagnostics in CNS tumor classification. At the same time, it remains wedded to other established approaches to tumor diagnosis such as histology and immunohistochemistry. In doing so, the fifth edition establishes some different approaches to both CNS tumor nomenclature and grading and it emphasizes the importance of integrated diagnoses and layered reports. New tumor types and subtypes are introduced, some based on novel diagnostic technologies such as DNA methylome profiling. The present review summarizes the major general changes in the 2021 fifth edition classification and the specific changes in each taxonomic category. It is hoped that this summary provides an overview to facilitate more in-depth exploration of the entire fifth edition of the WHO Classification of Tumors of the Central Nervous System.
The fifth edition of the WHO Classification of Tumors of the Central Nervous System (WHO CNS5)1 is the sixth version of the international standard for the classification of brain and spinal cord tumors, following the prior publications from 1979, 1993, 2000, 2007, and 2016.2–6 WHO CNS5 builds on the updated fourth edition that appeared in 2016, on the many developments in the field that followed the 2016 classification, and on the recommendations of the Consortium to Inform Molecular and Practical Approaches to CNS Tumor Taxonomy (cIMPACT-NOW).7–16 WHO CNS5 features substantial changes by moving further to advance the role of molecular diagnostics in CNS tumor classification but remaining rooted in other established approaches to tumor characterization, including histology and immunohistochemistry. WHO CNS5 is presented in Table 1, and the major general and specific changes are summarized in this review.
2021 WHO Classification of Tumors of the Central Nervous System. Provisional Entities are in Italics
| World Health Organization Classification of Tumors of the Central Nervous System, fifth edition |
|---|
| Gliomas, glioneuronal tumors, and neuronal tumors |
| Adult-type diffuse gliomas |
| Astrocytoma, IDH-mutant |
| Oligodendroglioma, IDH-mutant, and 1p/19q-codeleted |
| Glioblastoma, IDH-wildtype |
| Pediatric-type diffuse low-grade gliomas |
| Diffuse astrocytoma, MYB- or MYBL1-altered |
| Angiocentric glioma |
| Polymorphous low-grade neuroepithelial tumor of the young |
| Diffuse low-grade glioma, MAPK pathway-altered |
| Pediatric-type diffuse high-grade gliomas |
| Diffuse midline glioma, H3 K27-altered |
| Diffuse hemispheric glioma, H3 G34-mutant |
| Diffuse pediatric-type high-grade glioma, H3-wildtype and IDH-wildtype |
| Infant-type hemispheric glioma |
| Circumscribed astrocytic gliomas |
| Pilocytic astrocytoma |
| High-grade astrocytoma with piloid features |
| Pleomorphic xanthoastrocytoma |
| Subependymal giant cell astrocytoma |
| Chordoid glioma |
| Astroblastoma, MN1-altered |
| Glioneuronal and neuronal tumors |
| Ganglioglioma |
| Desmoplastic infantile ganglioglioma / desmoplastic infantile astrocytoma |
| Dysembryoplastic neuroepithelial tumor |
| Diffuse glioneuronal tumor with oligodendroglioma-like features and nuclear clusters |
| Papillary glioneuronal tumor |
| Rosette-forming glioneuronal tumor |
| Myxoid glioneuronal tumor |
| Diffuse leptomeningeal glioneuronal tumor |
| Gangliocytoma |
| Multinodular and vacuolating neuronal tumor |
| Dysplastic cerebellar gangliocytoma (Lhermitte-Duclos disease) |
| Central neurocytoma |
| Extraventricular neurocytoma |
| Cerebellar liponeurocytoma |
| Ependymal tumors |
| Supratentorial ependymoma |
| Supratentorial ependymoma, ZFTA fusion-positive |
| Supratentorial ependymoma, YAP1 fusion-positive |
| Posterior fossa ependymoma |
| Posterior fossa ependymoma, group PFA |
| Posterior fossa ependymoma, group PFB |
| Spinal ependymoma |
| Spinal ependymoma, MYCN-amplified |
| Myxopapillary ependymoma |
| Subependymoma |
| Choroid plexus tumors |
| Choroid plexus papilloma |
| Atypical choroid plexus papilloma |
| Choroid plexus carcinoma |
| Embryonal tumors |
| Medulloblastoma |
| Medulloblastomas, molecularly defined |
| Medulloblastoma, WNT-activated |
| Medulloblastoma, SHH-activated and TP53-wildtype |
| Medulloblastoma, SHH-activated and TP53-mutant |
| Medulloblastoma, non-WNT/non-SHH |
| Medulloblastomas, histologically defined |
| Other CNS embryonal tumors |
| Atypical teratoid/rhabdoid tumor |
| Cribriform neuroepithelial tumor |
| Embryonal tumor with multilayered rosettes |
| CNS neuroblastoma, FOXR2-activated |
| CNS tumor with BCOR internal tandem duplication |
| CNS embryonal tumor |
| Pineal tumors |
| Pineocytoma |
| Pineal parenchymal tumor of intermediate differentiation |
| Pineoblastoma |
| Papillary tumor of the pineal region |
| Desmoplastic myxoid tumor of the pineal region, SMARCB1-mutant |
| Cranial and paraspinal nerve tumors |
| Schwannoma |
| Neurofibroma |
| Perineurioma |
| Hybrid nerve sheath tumor |
| Malignant melanotic nerve sheath tumor |
| Malignant peripheral nerve sheath tumor |
| Paraganglioma |
| Meningiomas |
| Meningioma |
| Mesenchymal, non-meningothelial tumors |
| Soft tissue tumors |
| Fibroblastic and myofibroblastic tumors |
| Solitary fibrous tumor |
| Vascular tumors |
| Hemangiomas and vascular malformations |
| Hemangioblastoma |
| Skeletal muscle tumors |
| Rhabdomyosarcoma |
| Uncertain differentiation |
| Intracranial mesenchymal tumor, FET-CREB fusion-positive |
| CIC-rearranged sarcoma |
| Primary intracranial sarcoma, DICER1-mutant |
| Ewing sarcoma |
| Chondro-osseous tumors |
| Chondrogenic tumors |
| Mesenchymal chondrosarcoma |
| Chondrosarcoma |
| Notochordal tumors |
| Chordoma (including poorly differentiated chordoma) |
| Melanocytic tumors |
| Diffuse meningeal melanocytic neoplasms |
| Meningeal melanocytosis and meningeal melanomatosis |
| Circumscribed meningeal melanocytic neoplasms |
| Meningeal melanocytoma and meningeal melanoma |
| Hematolymphoid tumors |
| Lymphomas |
| CNS lymphomas |
| Primary diffuse large B-cell lymphoma of the CNS |
| Immunodeficiency-associated CNS lymphoma |
| Lymphomatoid granulomatosis |
| Intravascular large B-cell lymphoma |
| Miscellaneous rare lymphomas in the CNS |
| MALT lymphoma of the dura |
| Other low-grade B-cell lymphomas of the CNS |
| Anaplastic large cell lymphoma (ALK+/ALK−) |
| T-cell and NK/T-cell lymphomas |
| Histiocytic tumors |
| Erdheim-Chester disease |
| Rosai-Dorfman disease |
| Juvenile xanthogranuloma |
| Langerhans cell histiocytosis |
| Histiocytic sarcoma |
| Germ cell tumors |
| Mature teratoma |
| Immature teratoma |
| Teratoma with somatic-type malignancy |
| Germinoma |
| Embryonal carcinoma |
| Yolk sac tumor |
| Choriocarcinoma |
| Mixed germ cell tumor |
| Tumors of the sellar region |
| Adamantinomatous craniopharyngioma |
| Papillary craniopharyngioma |
| Pituicytoma, granular cell tumor of the sellar region, and spindle cell oncocytoma |
| Pituitary adenoma/PitNET |
| Pituitary blastoma |
| Metastases to the CNS |
| Metastases to the brain and spinal cord parenchyma |
| Metastases to the meninges |
| World Health Organization Classification of Tumors of the Central Nervous System, fifth edition |
|---|
| Gliomas, glioneuronal tumors, and neuronal tumors |
| Adult-type diffuse gliomas |
| Astrocytoma, IDH-mutant |
| Oligodendroglioma, IDH-mutant, and 1p/19q-codeleted |
| Glioblastoma, IDH-wildtype |
| Pediatric-type diffuse low-grade gliomas |
| Diffuse astrocytoma, MYB- or MYBL1-altered |
| Angiocentric glioma |
| Polymorphous low-grade neuroepithelial tumor of the young |
| Diffuse low-grade glioma, MAPK pathway-altered |
| Pediatric-type diffuse high-grade gliomas |
| Diffuse midline glioma, H3 K27-altered |
| Diffuse hemispheric glioma, H3 G34-mutant |
| Diffuse pediatric-type high-grade glioma, H3-wildtype and IDH-wildtype |
| Infant-type hemispheric glioma |
| Circumscribed astrocytic gliomas |
| Pilocytic astrocytoma |
| High-grade astrocytoma with piloid features |
| Pleomorphic xanthoastrocytoma |
| Subependymal giant cell astrocytoma |
| Chordoid glioma |
| Astroblastoma, MN1-altered |
| Glioneuronal and neuronal tumors |
| Ganglioglioma |
| Desmoplastic infantile ganglioglioma / desmoplastic infantile astrocytoma |
| Dysembryoplastic neuroepithelial tumor |
| Diffuse glioneuronal tumor with oligodendroglioma-like features and nuclear clusters |
| Papillary glioneuronal tumor |
| Rosette-forming glioneuronal tumor |
| Myxoid glioneuronal tumor |
| Diffuse leptomeningeal glioneuronal tumor |
| Gangliocytoma |
| Multinodular and vacuolating neuronal tumor |
| Dysplastic cerebellar gangliocytoma (Lhermitte-Duclos disease) |
| Central neurocytoma |
| Extraventricular neurocytoma |
| Cerebellar liponeurocytoma |
| Ependymal tumors |
| Supratentorial ependymoma |
| Supratentorial ependymoma, ZFTA fusion-positive |
| Supratentorial ependymoma, YAP1 fusion-positive |
| Posterior fossa ependymoma |
| Posterior fossa ependymoma, group PFA |
| Posterior fossa ependymoma, group PFB |
| Spinal ependymoma |
| Spinal ependymoma, MYCN-amplified |
| Myxopapillary ependymoma |
| Subependymoma |
| Choroid plexus tumors |
| Choroid plexus papilloma |
| Atypical choroid plexus papilloma |
| Choroid plexus carcinoma |
| Embryonal tumors |
| Medulloblastoma |
| Medulloblastomas, molecularly defined |
| Medulloblastoma, WNT-activated |
| Medulloblastoma, SHH-activated and TP53-wildtype |
| Medulloblastoma, SHH-activated and TP53-mutant |
| Medulloblastoma, non-WNT/non-SHH |
| Medulloblastomas, histologically defined |
| Other CNS embryonal tumors |
| Atypical teratoid/rhabdoid tumor |
| Cribriform neuroepithelial tumor |
| Embryonal tumor with multilayered rosettes |
| CNS neuroblastoma, FOXR2-activated |
| CNS tumor with BCOR internal tandem duplication |
| CNS embryonal tumor |
| Pineal tumors |
| Pineocytoma |
| Pineal parenchymal tumor of intermediate differentiation |
| Pineoblastoma |
| Papillary tumor of the pineal region |
| Desmoplastic myxoid tumor of the pineal region, SMARCB1-mutant |
| Cranial and paraspinal nerve tumors |
| Schwannoma |
| Neurofibroma |
| Perineurioma |
| Hybrid nerve sheath tumor |
| Malignant melanotic nerve sheath tumor |
| Malignant peripheral nerve sheath tumor |
| Paraganglioma |
| Meningiomas |
| Meningioma |
| Mesenchymal, non-meningothelial tumors |
| Soft tissue tumors |
| Fibroblastic and myofibroblastic tumors |
| Solitary fibrous tumor |
| Vascular tumors |
| Hemangiomas and vascular malformations |
| Hemangioblastoma |
| Skeletal muscle tumors |
| Rhabdomyosarcoma |
| Uncertain differentiation |
| Intracranial mesenchymal tumor, FET-CREB fusion-positive |
| CIC-rearranged sarcoma |
| Primary intracranial sarcoma, DICER1-mutant |
| Ewing sarcoma |
| Chondro-osseous tumors |
| Chondrogenic tumors |
| Mesenchymal chondrosarcoma |
| Chondrosarcoma |
| Notochordal tumors |
| Chordoma (including poorly differentiated chordoma) |
| Melanocytic tumors |
| Diffuse meningeal melanocytic neoplasms |
| Meningeal melanocytosis and meningeal melanomatosis |
| Circumscribed meningeal melanocytic neoplasms |
| Meningeal melanocytoma and meningeal melanoma |
| Hematolymphoid tumors |
| Lymphomas |
| CNS lymphomas |
| Primary diffuse large B-cell lymphoma of the CNS |
| Immunodeficiency-associated CNS lymphoma |
| Lymphomatoid granulomatosis |
| Intravascular large B-cell lymphoma |
| Miscellaneous rare lymphomas in the CNS |
| MALT lymphoma of the dura |
| Other low-grade B-cell lymphomas of the CNS |
| Anaplastic large cell lymphoma (ALK+/ALK−) |
| T-cell and NK/T-cell lymphomas |
| Histiocytic tumors |
| Erdheim-Chester disease |
| Rosai-Dorfman disease |
| Juvenile xanthogranuloma |
| Langerhans cell histiocytosis |
| Histiocytic sarcoma |
| Germ cell tumors |
| Mature teratoma |
| Immature teratoma |
| Teratoma with somatic-type malignancy |
| Germinoma |
| Embryonal carcinoma |
| Yolk sac tumor |
| Choriocarcinoma |
| Mixed germ cell tumor |
| Tumors of the sellar region |
| Adamantinomatous craniopharyngioma |
| Papillary craniopharyngioma |
| Pituicytoma, granular cell tumor of the sellar region, and spindle cell oncocytoma |
| Pituitary adenoma/PitNET |
| Pituitary blastoma |
| Metastases to the CNS |
| Metastases to the brain and spinal cord parenchyma |
| Metastases to the meninges |
Abbreviations: CNS, central nervous system; IDH, isocitrate dehydrogenase; NK, natural killer; PitNET, pituitary neuroendocrine tumor; SHH, sonic hedgehog.
2021 WHO Classification of Tumors of the Central Nervous System. Provisional Entities are in Italics
| World Health Organization Classification of Tumors of the Central Nervous System, fifth edition |
|---|
| Gliomas, glioneuronal tumors, and neuronal tumors |
| Adult-type diffuse gliomas |
| Astrocytoma, IDH-mutant |
| Oligodendroglioma, IDH-mutant, and 1p/19q-codeleted |
| Glioblastoma, IDH-wildtype |
| Pediatric-type diffuse low-grade gliomas |
| Diffuse astrocytoma, MYB- or MYBL1-altered |
| Angiocentric glioma |
| Polymorphous low-grade neuroepithelial tumor of the young |
| Diffuse low-grade glioma, MAPK pathway-altered |
| Pediatric-type diffuse high-grade gliomas |
| Diffuse midline glioma, H3 K27-altered |
| Diffuse hemispheric glioma, H3 G34-mutant |
| Diffuse pediatric-type high-grade glioma, H3-wildtype and IDH-wildtype |
| Infant-type hemispheric glioma |
| Circumscribed astrocytic gliomas |
| Pilocytic astrocytoma |
| High-grade astrocytoma with piloid features |
| Pleomorphic xanthoastrocytoma |
| Subependymal giant cell astrocytoma |
| Chordoid glioma |
| Astroblastoma, MN1-altered |
| Glioneuronal and neuronal tumors |
| Ganglioglioma |
| Desmoplastic infantile ganglioglioma / desmoplastic infantile astrocytoma |
| Dysembryoplastic neuroepithelial tumor |
| Diffuse glioneuronal tumor with oligodendroglioma-like features and nuclear clusters |
| Papillary glioneuronal tumor |
| Rosette-forming glioneuronal tumor |
| Myxoid glioneuronal tumor |
| Diffuse leptomeningeal glioneuronal tumor |
| Gangliocytoma |
| Multinodular and vacuolating neuronal tumor |
| Dysplastic cerebellar gangliocytoma (Lhermitte-Duclos disease) |
| Central neurocytoma |
| Extraventricular neurocytoma |
| Cerebellar liponeurocytoma |
| Ependymal tumors |
| Supratentorial ependymoma |
| Supratentorial ependymoma, ZFTA fusion-positive |
| Supratentorial ependymoma, YAP1 fusion-positive |
| Posterior fossa ependymoma |
| Posterior fossa ependymoma, group PFA |
| Posterior fossa ependymoma, group PFB |
| Spinal ependymoma |
| Spinal ependymoma, MYCN-amplified |
| Myxopapillary ependymoma |
| Subependymoma |
| Choroid plexus tumors |
| Choroid plexus papilloma |
| Atypical choroid plexus papilloma |
| Choroid plexus carcinoma |
| Embryonal tumors |
| Medulloblastoma |
| Medulloblastomas, molecularly defined |
| Medulloblastoma, WNT-activated |
| Medulloblastoma, SHH-activated and TP53-wildtype |
| Medulloblastoma, SHH-activated and TP53-mutant |
| Medulloblastoma, non-WNT/non-SHH |
| Medulloblastomas, histologically defined |
| Other CNS embryonal tumors |
| Atypical teratoid/rhabdoid tumor |
| Cribriform neuroepithelial tumor |
| Embryonal tumor with multilayered rosettes |
| CNS neuroblastoma, FOXR2-activated |
| CNS tumor with BCOR internal tandem duplication |
| CNS embryonal tumor |
| Pineal tumors |
| Pineocytoma |
| Pineal parenchymal tumor of intermediate differentiation |
| Pineoblastoma |
| Papillary tumor of the pineal region |
| Desmoplastic myxoid tumor of the pineal region, SMARCB1-mutant |
| Cranial and paraspinal nerve tumors |
| Schwannoma |
| Neurofibroma |
| Perineurioma |
| Hybrid nerve sheath tumor |
| Malignant melanotic nerve sheath tumor |
| Malignant peripheral nerve sheath tumor |
| Paraganglioma |
| Meningiomas |
| Meningioma |
| Mesenchymal, non-meningothelial tumors |
| Soft tissue tumors |
| Fibroblastic and myofibroblastic tumors |
| Solitary fibrous tumor |
| Vascular tumors |
| Hemangiomas and vascular malformations |
| Hemangioblastoma |
| Skeletal muscle tumors |
| Rhabdomyosarcoma |
| Uncertain differentiation |
| Intracranial mesenchymal tumor, FET-CREB fusion-positive |
| CIC-rearranged sarcoma |
| Primary intracranial sarcoma, DICER1-mutant |
| Ewing sarcoma |
| Chondro-osseous tumors |
| Chondrogenic tumors |
| Mesenchymal chondrosarcoma |
| Chondrosarcoma |
| Notochordal tumors |
| Chordoma (including poorly differentiated chordoma) |
| Melanocytic tumors |
| Diffuse meningeal melanocytic neoplasms |
| Meningeal melanocytosis and meningeal melanomatosis |
| Circumscribed meningeal melanocytic neoplasms |
| Meningeal melanocytoma and meningeal melanoma |
| Hematolymphoid tumors |
| Lymphomas |
| CNS lymphomas |
| Primary diffuse large B-cell lymphoma of the CNS |
| Immunodeficiency-associated CNS lymphoma |
| Lymphomatoid granulomatosis |
| Intravascular large B-cell lymphoma |
| Miscellaneous rare lymphomas in the CNS |
| MALT lymphoma of the dura |
| Other low-grade B-cell lymphomas of the CNS |
| Anaplastic large cell lymphoma (ALK+/ALK−) |
| T-cell and NK/T-cell lymphomas |
| Histiocytic tumors |
| Erdheim-Chester disease |
| Rosai-Dorfman disease |
| Juvenile xanthogranuloma |
| Langerhans cell histiocytosis |
| Histiocytic sarcoma |
| Germ cell tumors |
| Mature teratoma |
| Immature teratoma |
| Teratoma with somatic-type malignancy |
| Germinoma |
| Embryonal carcinoma |
| Yolk sac tumor |
| Choriocarcinoma |
| Mixed germ cell tumor |
| Tumors of the sellar region |
| Adamantinomatous craniopharyngioma |
| Papillary craniopharyngioma |
| Pituicytoma, granular cell tumor of the sellar region, and spindle cell oncocytoma |
| Pituitary adenoma/PitNET |
| Pituitary blastoma |
| Metastases to the CNS |
| Metastases to the brain and spinal cord parenchyma |
| Metastases to the meninges |
| World Health Organization Classification of Tumors of the Central Nervous System, fifth edition |
|---|
| Gliomas, glioneuronal tumors, and neuronal tumors |
| Adult-type diffuse gliomas |
| Astrocytoma, IDH-mutant |
| Oligodendroglioma, IDH-mutant, and 1p/19q-codeleted |
| Glioblastoma, IDH-wildtype |
| Pediatric-type diffuse low-grade gliomas |
| Diffuse astrocytoma, MYB- or MYBL1-altered |
| Angiocentric glioma |
| Polymorphous low-grade neuroepithelial tumor of the young |
| Diffuse low-grade glioma, MAPK pathway-altered |
| Pediatric-type diffuse high-grade gliomas |
| Diffuse midline glioma, H3 K27-altered |
| Diffuse hemispheric glioma, H3 G34-mutant |
| Diffuse pediatric-type high-grade glioma, H3-wildtype and IDH-wildtype |
| Infant-type hemispheric glioma |
| Circumscribed astrocytic gliomas |
| Pilocytic astrocytoma |
| High-grade astrocytoma with piloid features |
| Pleomorphic xanthoastrocytoma |
| Subependymal giant cell astrocytoma |
| Chordoid glioma |
| Astroblastoma, MN1-altered |
| Glioneuronal and neuronal tumors |
| Ganglioglioma |
| Desmoplastic infantile ganglioglioma / desmoplastic infantile astrocytoma |
| Dysembryoplastic neuroepithelial tumor |
| Diffuse glioneuronal tumor with oligodendroglioma-like features and nuclear clusters |
| Papillary glioneuronal tumor |
| Rosette-forming glioneuronal tumor |
| Myxoid glioneuronal tumor |
| Diffuse leptomeningeal glioneuronal tumor |
| Gangliocytoma |
| Multinodular and vacuolating neuronal tumor |
| Dysplastic cerebellar gangliocytoma (Lhermitte-Duclos disease) |
| Central neurocytoma |
| Extraventricular neurocytoma |
| Cerebellar liponeurocytoma |
| Ependymal tumors |
| Supratentorial ependymoma |
| Supratentorial ependymoma, ZFTA fusion-positive |
| Supratentorial ependymoma, YAP1 fusion-positive |
| Posterior fossa ependymoma |
| Posterior fossa ependymoma, group PFA |
| Posterior fossa ependymoma, group PFB |
| Spinal ependymoma |
| Spinal ependymoma, MYCN-amplified |
| Myxopapillary ependymoma |
| Subependymoma |
| Choroid plexus tumors |
| Choroid plexus papilloma |
| Atypical choroid plexus papilloma |
| Choroid plexus carcinoma |
| Embryonal tumors |
| Medulloblastoma |
| Medulloblastomas, molecularly defined |
| Medulloblastoma, WNT-activated |
| Medulloblastoma, SHH-activated and TP53-wildtype |
| Medulloblastoma, SHH-activated and TP53-mutant |
| Medulloblastoma, non-WNT/non-SHH |
| Medulloblastomas, histologically defined |
| Other CNS embryonal tumors |
| Atypical teratoid/rhabdoid tumor |
| Cribriform neuroepithelial tumor |
| Embryonal tumor with multilayered rosettes |
| CNS neuroblastoma, FOXR2-activated |
| CNS tumor with BCOR internal tandem duplication |
| CNS embryonal tumor |
| Pineal tumors |
| Pineocytoma |
| Pineal parenchymal tumor of intermediate differentiation |
| Pineoblastoma |
| Papillary tumor of the pineal region |
| Desmoplastic myxoid tumor of the pineal region, SMARCB1-mutant |
| Cranial and paraspinal nerve tumors |
| Schwannoma |
| Neurofibroma |
| Perineurioma |
| Hybrid nerve sheath tumor |
| Malignant melanotic nerve sheath tumor |
| Malignant peripheral nerve sheath tumor |
| Paraganglioma |
| Meningiomas |
| Meningioma |
| Mesenchymal, non-meningothelial tumors |
| Soft tissue tumors |
| Fibroblastic and myofibroblastic tumors |
| Solitary fibrous tumor |
| Vascular tumors |
| Hemangiomas and vascular malformations |
| Hemangioblastoma |
| Skeletal muscle tumors |
| Rhabdomyosarcoma |
| Uncertain differentiation |
| Intracranial mesenchymal tumor, FET-CREB fusion-positive |
| CIC-rearranged sarcoma |
| Primary intracranial sarcoma, DICER1-mutant |
| Ewing sarcoma |
| Chondro-osseous tumors |
| Chondrogenic tumors |
| Mesenchymal chondrosarcoma |
| Chondrosarcoma |
| Notochordal tumors |
| Chordoma (including poorly differentiated chordoma) |
| Melanocytic tumors |
| Diffuse meningeal melanocytic neoplasms |
| Meningeal melanocytosis and meningeal melanomatosis |
| Circumscribed meningeal melanocytic neoplasms |
| Meningeal melanocytoma and meningeal melanoma |
| Hematolymphoid tumors |
| Lymphomas |
| CNS lymphomas |
| Primary diffuse large B-cell lymphoma of the CNS |
| Immunodeficiency-associated CNS lymphoma |
| Lymphomatoid granulomatosis |
| Intravascular large B-cell lymphoma |
| Miscellaneous rare lymphomas in the CNS |
| MALT lymphoma of the dura |
| Other low-grade B-cell lymphomas of the CNS |
| Anaplastic large cell lymphoma (ALK+/ALK−) |
| T-cell and NK/T-cell lymphomas |
| Histiocytic tumors |
| Erdheim-Chester disease |
| Rosai-Dorfman disease |
| Juvenile xanthogranuloma |
| Langerhans cell histiocytosis |
| Histiocytic sarcoma |
| Germ cell tumors |
| Mature teratoma |
| Immature teratoma |
| Teratoma with somatic-type malignancy |
| Germinoma |
| Embryonal carcinoma |
| Yolk sac tumor |
| Choriocarcinoma |
| Mixed germ cell tumor |
| Tumors of the sellar region |
| Adamantinomatous craniopharyngioma |
| Papillary craniopharyngioma |
| Pituicytoma, granular cell tumor of the sellar region, and spindle cell oncocytoma |
| Pituitary adenoma/PitNET |
| Pituitary blastoma |
| Metastases to the CNS |
| Metastases to the brain and spinal cord parenchyma |
| Metastases to the meninges |
Abbreviations: CNS, central nervous system; IDH, isocitrate dehydrogenase; NK, natural killer; PitNET, pituitary neuroendocrine tumor; SHH, sonic hedgehog.
General Changes
CNS Tumor Taxonomy
CNS tumor classification has long been based on histological findings supported by ancillary tissue-based tests (eg, immunohistochemical, ultrastructural). More recently, molecular biomarkers have gained importance in providing both ancillary and defining diagnostic information. WHO CNS5 therefore incorporates numerous molecular changes with clinicopathologic utility that are important for the most accurate classification of CNS neoplasms. Table 2 catalogs the key genes and proteins that are analyzed for diagnostic alterations important for integrated CNS tumor classification. WHO CNS5 does not recommend specific methods for molecular assessment of the individual diagnostic alterations unless a certain method is unequivocally required for the diagnosis of a distinct tumor type or subtype (see below).
Key Diagnostic Genes, Molecules, Pathways, and/or Combinations in Major Primary CNS Tumors
| Tumor Type | Genes/Molecular Profiles Characteristically Altereda |
|---|---|
| Astrocytoma, IDH-mutant | IDH1, IDH2, ATRX, TP53, CDKN2A/B |
| Oligodendroglioma, IDH-mutant, and 1p/19q-codeleted | IDH1, IDH2, 1p/19q, TERT promoter, CIC, FUBP1, NOTCH1 |
| Glioblastoma, IDH-wildtype | IDH-wildtype, TERT promoter, chromosomes 7/10, EGFR |
| Diffuse astrocytoma, MYB- or MYBL1-altered | MYB, MYBL1 |
| Angiocentric glioma | MYB |
| Polymorphous low-grade neuroepithelial tumor of the young | BRAF, FGFR family |
| Diffuse low-grade glioma, MAPK pathway-altered | FGFR1, BRAF |
| Diffuse midline glioma, H3 K27-altered | H3 K27, TP53, ACVR1, PDGFRA, EGFR, EZHIP |
| Diffuse hemispheric glioma, H3 G34-mutant | H3 G34, TP53, ATRX |
| Diffuse pediatric-type high-grade glioma, H3-wildtype, and IDH-wildtype | IDH-wildtype, H3-wildtype, PDGFRA, MYCN, EGFR (methylome) |
| Infant-type hemispheric glioma | NTRK family, ALK, ROS, MET |
| Pilocytic astrocytoma | KIAA1549-BRAF, BRAF, NF1 |
| High-grade astrocytoma with piloid features | BRAF, NF1, ATRX, CDKN2A/B (methylome) |
| Pleomorphic xanthoastrocytoma | BRAF, CDKN2A/B |
| Subependymal giant cell astrocytoma | TSC1, TSC2 |
| Chordoid glioma | PRKCA |
| Astroblastoma, MN1-altered | MN1 |
| Ganglion cell tumors | BRAF |
| Dysembryoplastic neuroepithelial tumor | FGFR1 |
| Diffuse glioneuronal tumor with oligodendroglioma-like features and nuclear clusters | Chromosome 14, (methylome) |
| Papillary glioneuronal tumor | PRKCA |
| Rosette-forming glioneuronal tumor | FGFR1, PIK3CA, NF1 |
| Myxoid glioneuronal tumor | PDFGRA |
| Diffuse leptomeningeal glioneuronal tumor | KIAA1549-BRAF fusion, 1p (methylome) |
| Multinodular and vacuolating neuronal tumor | MAPK pathway |
| Dysplastic cerebellar gangliocytoma (Lhermitte-Duclos disease) | PTEN |
| Extraventricular neurocytoma | FGFR (FGFR1-TACC1 fusion), IDH-wildtype |
| Supratentorial ependymomas | ZFTA, RELA, YAP1, MAML2 |
| Posterior fossa ependymomas | H3 K27me3, EZHIP (methylome) |
| Spinal ependymomas | NF2, MYCN |
| Medulloblastoma, WNT-activated | CTNNB1, APC |
| Medulloblastoma, SHH-activated | TP53, PTCH1, SUFU, SMO, MYCN, GLI2 (methylome) |
| Medulloblastoma, non-WNT/non-SHH | MYC, MYCN, PRDM6, KDM6A (methylome) |
| Atypical teratoid/rhabdoid tumor | SMARCB1, SMARCA4 |
| Embryonal tumor with multilayered rosettes | C19MC, DICER1 |
| CNS neuroblastoma, FOXR2-activated | FOXR2 |
| CNS tumor with BCOR internal tandem duplication | BCOR |
| Desmoplastic myxoid tumor of the pineal region, SMARCB1-mutant | SMARCB1 |
| Meningiomas | NF2, AKT1, TRAF7, SMO, PIK3CA; KLF4, SMARCE1, BAP1 in subtypes; H3K27me3; TERT promoter, CDKN2A/B in CNS WHO grade 3 |
| Solitary fibrous tumor | NAB2-STAT6 |
| Meningeal melanocytic tumors | NRAS (diffuse); GNAQ, GNA11, PLCB4, CYSLTR2 (circumscribed) |
| Adamantinomatous craniopharyngioma | CTNNB1 |
| Papillary craniopharyngioma | BRAF |
| Tumor Type | Genes/Molecular Profiles Characteristically Altereda |
|---|---|
| Astrocytoma, IDH-mutant | IDH1, IDH2, ATRX, TP53, CDKN2A/B |
| Oligodendroglioma, IDH-mutant, and 1p/19q-codeleted | IDH1, IDH2, 1p/19q, TERT promoter, CIC, FUBP1, NOTCH1 |
| Glioblastoma, IDH-wildtype | IDH-wildtype, TERT promoter, chromosomes 7/10, EGFR |
| Diffuse astrocytoma, MYB- or MYBL1-altered | MYB, MYBL1 |
| Angiocentric glioma | MYB |
| Polymorphous low-grade neuroepithelial tumor of the young | BRAF, FGFR family |
| Diffuse low-grade glioma, MAPK pathway-altered | FGFR1, BRAF |
| Diffuse midline glioma, H3 K27-altered | H3 K27, TP53, ACVR1, PDGFRA, EGFR, EZHIP |
| Diffuse hemispheric glioma, H3 G34-mutant | H3 G34, TP53, ATRX |
| Diffuse pediatric-type high-grade glioma, H3-wildtype, and IDH-wildtype | IDH-wildtype, H3-wildtype, PDGFRA, MYCN, EGFR (methylome) |
| Infant-type hemispheric glioma | NTRK family, ALK, ROS, MET |
| Pilocytic astrocytoma | KIAA1549-BRAF, BRAF, NF1 |
| High-grade astrocytoma with piloid features | BRAF, NF1, ATRX, CDKN2A/B (methylome) |
| Pleomorphic xanthoastrocytoma | BRAF, CDKN2A/B |
| Subependymal giant cell astrocytoma | TSC1, TSC2 |
| Chordoid glioma | PRKCA |
| Astroblastoma, MN1-altered | MN1 |
| Ganglion cell tumors | BRAF |
| Dysembryoplastic neuroepithelial tumor | FGFR1 |
| Diffuse glioneuronal tumor with oligodendroglioma-like features and nuclear clusters | Chromosome 14, (methylome) |
| Papillary glioneuronal tumor | PRKCA |
| Rosette-forming glioneuronal tumor | FGFR1, PIK3CA, NF1 |
| Myxoid glioneuronal tumor | PDFGRA |
| Diffuse leptomeningeal glioneuronal tumor | KIAA1549-BRAF fusion, 1p (methylome) |
| Multinodular and vacuolating neuronal tumor | MAPK pathway |
| Dysplastic cerebellar gangliocytoma (Lhermitte-Duclos disease) | PTEN |
| Extraventricular neurocytoma | FGFR (FGFR1-TACC1 fusion), IDH-wildtype |
| Supratentorial ependymomas | ZFTA, RELA, YAP1, MAML2 |
| Posterior fossa ependymomas | H3 K27me3, EZHIP (methylome) |
| Spinal ependymomas | NF2, MYCN |
| Medulloblastoma, WNT-activated | CTNNB1, APC |
| Medulloblastoma, SHH-activated | TP53, PTCH1, SUFU, SMO, MYCN, GLI2 (methylome) |
| Medulloblastoma, non-WNT/non-SHH | MYC, MYCN, PRDM6, KDM6A (methylome) |
| Atypical teratoid/rhabdoid tumor | SMARCB1, SMARCA4 |
| Embryonal tumor with multilayered rosettes | C19MC, DICER1 |
| CNS neuroblastoma, FOXR2-activated | FOXR2 |
| CNS tumor with BCOR internal tandem duplication | BCOR |
| Desmoplastic myxoid tumor of the pineal region, SMARCB1-mutant | SMARCB1 |
| Meningiomas | NF2, AKT1, TRAF7, SMO, PIK3CA; KLF4, SMARCE1, BAP1 in subtypes; H3K27me3; TERT promoter, CDKN2A/B in CNS WHO grade 3 |
| Solitary fibrous tumor | NAB2-STAT6 |
| Meningeal melanocytic tumors | NRAS (diffuse); GNAQ, GNA11, PLCB4, CYSLTR2 (circumscribed) |
| Adamantinomatous craniopharyngioma | CTNNB1 |
| Papillary craniopharyngioma | BRAF |
Abbreviations: CNS, central nervous system; C19MC, chromosome 19 microRNA cluster; IDH, isocitrate dehydrogenase; SHH, sonic hedgehog.
Some of these are definitional for specific diagnoses, while others are not definitional but are characteristically altered or not altered. For each tumor type, these distinctions are specified in the Diagnostic Molecular Pathology as well as the Essential and Desirable Criteria sections of the Blue Book chapters.
aIn this column, molecules that are definitional (including for those that are wildtype) are listed before others; for those tumor types without specific definitional changes, more commonly altered genes and molecules are listed before others. Most types have characteristic methylome patterns, but “(methylome)” is only listed for those types for which methylome testing offers particular diagnostic guidance, including for designating subtypes (as for Medulloblastoma, SHH-activated; Medulloblastoma, non-WNT/non-SHH; and Diffuse leptomeningeal glioneuronal tumor). H3 is a gene family (eg, H3F3A, HIST1H3B).
Key Diagnostic Genes, Molecules, Pathways, and/or Combinations in Major Primary CNS Tumors
| Tumor Type | Genes/Molecular Profiles Characteristically Altereda |
|---|---|
| Astrocytoma, IDH-mutant | IDH1, IDH2, ATRX, TP53, CDKN2A/B |
| Oligodendroglioma, IDH-mutant, and 1p/19q-codeleted | IDH1, IDH2, 1p/19q, TERT promoter, CIC, FUBP1, NOTCH1 |
| Glioblastoma, IDH-wildtype | IDH-wildtype, TERT promoter, chromosomes 7/10, EGFR |
| Diffuse astrocytoma, MYB- or MYBL1-altered | MYB, MYBL1 |
| Angiocentric glioma | MYB |
| Polymorphous low-grade neuroepithelial tumor of the young | BRAF, FGFR family |
| Diffuse low-grade glioma, MAPK pathway-altered | FGFR1, BRAF |
| Diffuse midline glioma, H3 K27-altered | H3 K27, TP53, ACVR1, PDGFRA, EGFR, EZHIP |
| Diffuse hemispheric glioma, H3 G34-mutant | H3 G34, TP53, ATRX |
| Diffuse pediatric-type high-grade glioma, H3-wildtype, and IDH-wildtype | IDH-wildtype, H3-wildtype, PDGFRA, MYCN, EGFR (methylome) |
| Infant-type hemispheric glioma | NTRK family, ALK, ROS, MET |
| Pilocytic astrocytoma | KIAA1549-BRAF, BRAF, NF1 |
| High-grade astrocytoma with piloid features | BRAF, NF1, ATRX, CDKN2A/B (methylome) |
| Pleomorphic xanthoastrocytoma | BRAF, CDKN2A/B |
| Subependymal giant cell astrocytoma | TSC1, TSC2 |
| Chordoid glioma | PRKCA |
| Astroblastoma, MN1-altered | MN1 |
| Ganglion cell tumors | BRAF |
| Dysembryoplastic neuroepithelial tumor | FGFR1 |
| Diffuse glioneuronal tumor with oligodendroglioma-like features and nuclear clusters | Chromosome 14, (methylome) |
| Papillary glioneuronal tumor | PRKCA |
| Rosette-forming glioneuronal tumor | FGFR1, PIK3CA, NF1 |
| Myxoid glioneuronal tumor | PDFGRA |
| Diffuse leptomeningeal glioneuronal tumor | KIAA1549-BRAF fusion, 1p (methylome) |
| Multinodular and vacuolating neuronal tumor | MAPK pathway |
| Dysplastic cerebellar gangliocytoma (Lhermitte-Duclos disease) | PTEN |
| Extraventricular neurocytoma | FGFR (FGFR1-TACC1 fusion), IDH-wildtype |
| Supratentorial ependymomas | ZFTA, RELA, YAP1, MAML2 |
| Posterior fossa ependymomas | H3 K27me3, EZHIP (methylome) |
| Spinal ependymomas | NF2, MYCN |
| Medulloblastoma, WNT-activated | CTNNB1, APC |
| Medulloblastoma, SHH-activated | TP53, PTCH1, SUFU, SMO, MYCN, GLI2 (methylome) |
| Medulloblastoma, non-WNT/non-SHH | MYC, MYCN, PRDM6, KDM6A (methylome) |
| Atypical teratoid/rhabdoid tumor | SMARCB1, SMARCA4 |
| Embryonal tumor with multilayered rosettes | C19MC, DICER1 |
| CNS neuroblastoma, FOXR2-activated | FOXR2 |
| CNS tumor with BCOR internal tandem duplication | BCOR |
| Desmoplastic myxoid tumor of the pineal region, SMARCB1-mutant | SMARCB1 |
| Meningiomas | NF2, AKT1, TRAF7, SMO, PIK3CA; KLF4, SMARCE1, BAP1 in subtypes; H3K27me3; TERT promoter, CDKN2A/B in CNS WHO grade 3 |
| Solitary fibrous tumor | NAB2-STAT6 |
| Meningeal melanocytic tumors | NRAS (diffuse); GNAQ, GNA11, PLCB4, CYSLTR2 (circumscribed) |
| Adamantinomatous craniopharyngioma | CTNNB1 |
| Papillary craniopharyngioma | BRAF |
| Tumor Type | Genes/Molecular Profiles Characteristically Altereda |
|---|---|
| Astrocytoma, IDH-mutant | IDH1, IDH2, ATRX, TP53, CDKN2A/B |
| Oligodendroglioma, IDH-mutant, and 1p/19q-codeleted | IDH1, IDH2, 1p/19q, TERT promoter, CIC, FUBP1, NOTCH1 |
| Glioblastoma, IDH-wildtype | IDH-wildtype, TERT promoter, chromosomes 7/10, EGFR |
| Diffuse astrocytoma, MYB- or MYBL1-altered | MYB, MYBL1 |
| Angiocentric glioma | MYB |
| Polymorphous low-grade neuroepithelial tumor of the young | BRAF, FGFR family |
| Diffuse low-grade glioma, MAPK pathway-altered | FGFR1, BRAF |
| Diffuse midline glioma, H3 K27-altered | H3 K27, TP53, ACVR1, PDGFRA, EGFR, EZHIP |
| Diffuse hemispheric glioma, H3 G34-mutant | H3 G34, TP53, ATRX |
| Diffuse pediatric-type high-grade glioma, H3-wildtype, and IDH-wildtype | IDH-wildtype, H3-wildtype, PDGFRA, MYCN, EGFR (methylome) |
| Infant-type hemispheric glioma | NTRK family, ALK, ROS, MET |
| Pilocytic astrocytoma | KIAA1549-BRAF, BRAF, NF1 |
| High-grade astrocytoma with piloid features | BRAF, NF1, ATRX, CDKN2A/B (methylome) |
| Pleomorphic xanthoastrocytoma | BRAF, CDKN2A/B |
| Subependymal giant cell astrocytoma | TSC1, TSC2 |
| Chordoid glioma | PRKCA |
| Astroblastoma, MN1-altered | MN1 |
| Ganglion cell tumors | BRAF |
| Dysembryoplastic neuroepithelial tumor | FGFR1 |
| Diffuse glioneuronal tumor with oligodendroglioma-like features and nuclear clusters | Chromosome 14, (methylome) |
| Papillary glioneuronal tumor | PRKCA |
| Rosette-forming glioneuronal tumor | FGFR1, PIK3CA, NF1 |
| Myxoid glioneuronal tumor | PDFGRA |
| Diffuse leptomeningeal glioneuronal tumor | KIAA1549-BRAF fusion, 1p (methylome) |
| Multinodular and vacuolating neuronal tumor | MAPK pathway |
| Dysplastic cerebellar gangliocytoma (Lhermitte-Duclos disease) | PTEN |
| Extraventricular neurocytoma | FGFR (FGFR1-TACC1 fusion), IDH-wildtype |
| Supratentorial ependymomas | ZFTA, RELA, YAP1, MAML2 |
| Posterior fossa ependymomas | H3 K27me3, EZHIP (methylome) |
| Spinal ependymomas | NF2, MYCN |
| Medulloblastoma, WNT-activated | CTNNB1, APC |
| Medulloblastoma, SHH-activated | TP53, PTCH1, SUFU, SMO, MYCN, GLI2 (methylome) |
| Medulloblastoma, non-WNT/non-SHH | MYC, MYCN, PRDM6, KDM6A (methylome) |
| Atypical teratoid/rhabdoid tumor | SMARCB1, SMARCA4 |
| Embryonal tumor with multilayered rosettes | C19MC, DICER1 |
| CNS neuroblastoma, FOXR2-activated | FOXR2 |
| CNS tumor with BCOR internal tandem duplication | BCOR |
| Desmoplastic myxoid tumor of the pineal region, SMARCB1-mutant | SMARCB1 |
| Meningiomas | NF2, AKT1, TRAF7, SMO, PIK3CA; KLF4, SMARCE1, BAP1 in subtypes; H3K27me3; TERT promoter, CDKN2A/B in CNS WHO grade 3 |
| Solitary fibrous tumor | NAB2-STAT6 |
| Meningeal melanocytic tumors | NRAS (diffuse); GNAQ, GNA11, PLCB4, CYSLTR2 (circumscribed) |
| Adamantinomatous craniopharyngioma | CTNNB1 |
| Papillary craniopharyngioma | BRAF |
Abbreviations: CNS, central nervous system; C19MC, chromosome 19 microRNA cluster; IDH, isocitrate dehydrogenase; SHH, sonic hedgehog.
Some of these are definitional for specific diagnoses, while others are not definitional but are characteristically altered or not altered. For each tumor type, these distinctions are specified in the Diagnostic Molecular Pathology as well as the Essential and Desirable Criteria sections of the Blue Book chapters.
aIn this column, molecules that are definitional (including for those that are wildtype) are listed before others; for those tumor types without specific definitional changes, more commonly altered genes and molecules are listed before others. Most types have characteristic methylome patterns, but “(methylome)” is only listed for those types for which methylome testing offers particular diagnostic guidance, including for designating subtypes (as for Medulloblastoma, SHH-activated; Medulloblastoma, non-WNT/non-SHH; and Diffuse leptomeningeal glioneuronal tumor). H3 is a gene family (eg, H3F3A, HIST1H3B).
As the use of molecular biomarkers in brain and spinal cord tumor diagnosis has been further elucidated, challenges have arisen in how to organize the classification of tumor types. Some are readily and consistently characterized by defining molecular features; for some, molecular parameters are not required but may support their classification; yet others are rarely or never diagnosed using molecular approaches. The resulting nosological organization is therefore also mixed. For some tumor families, WHO CNS5 has grouped tumors according to the genetic changes that enable a complete diagnosis (eg, IDH and H3 status); by looser oncogenic associations, such as MAPK pathway alterations; by histological and histogenetic similarities even though molecular signatures vary (eg, see neoplasms listed under Other Gliomas, Glioneuronal Tumors, and Neuronal Tumors); or, for many, by using molecular features to define new types and subtypes (eg, medulloblastoma). This hybrid taxonomy represents the current state of the field but is likely only an intermediate stage to an even more precise future classification. Examples of such transitional states include tumor families, such as Pediatric-type diffuse low-grade gliomas, in which some tumor types encompass several subtypes with a shared molecular feature while other types are precisely defined by a single feature, with such consensus decisions being based on the state of the field at the time of final editorial discussions.
To standardize WHO CNS5 with other fifth-edition Blue Books, the term “type” is used instead of “entity” and “subtype” is used instead of “variant.” Only types are listed in the classification (Table 1), with subtypes listed in the Subtype(s) subsections and described under Histopathology and/or Diagnostic Molecular Pathology of individual sections. For example, as a result of this change and because grading is being applied within types (see below), Meningioma is a single type with only one entry in the classification, but with many histological subtypes and grades further described in the text.
CNS Tumor Nomenclature
For CNS tumor nomenclature, WHO CNS5 follows the recommendations of the 2019 cIMPACT-NOW Utrecht meeting to make nomenclature more consistent and simple.14 In the past, some tumor names had anatomic site modifiers (eg, Chordoid glioma of the third ventricle) whereas others did not, despite occurring in specific locations (eg, Medulloblastoma). Some included genetic modifiers (eg, Glioblastoma, IDH-wildtype), whereas others did not, despite having specific genotypes (eg, Atypical teratoid/rhabdoid tumor [AT/RT]). Names have therefore been simplified as much as possible, and only location, age, or genetic modifiers with clinical utility have been used (eg, Extraventricular neurocytoma vs Central neurocytoma). Importantly, for tumors with highly characteristic features (eg, that chordoid gliomas occur in the third ventricle), these are included in tumor definitions and descriptions, even if they are not part of a tumor name. In addition, tumor names sometimes reflect morphologic features that are not prominent in all examples of the type; for example, some myxopapillary ependymomas are minimally myxoid, and some may not be overtly papillary. Similarly, xanthomatous change may be limited to a small fraction of cells in pleomorphic xanthoastrocytomas. Nonetheless, such names represent characteristic, if not universal, features. The terms may also reflect historical associations that have become embedded in common usage; for instance, although a medulloblast has not been identified in developmental studies, the term medulloblastoma is deeply ingrained in tumor terminology, and changing the name could be quite disruptive to clinical care and scientific experiments that rely on prior data, as well as epidemiological studies. Lastly, with the change to grading within tumor type (see below), modifier terms like “anaplastic” are not routinely included; familiar names like “anaplastic astrocytoma” and “anaplastic oligodendroglioma” do not, therefore, appear in this classification.
Gene and Protein Nomenclature for CNS Tumor Classification
The fifth edition of the WHO Classification of Tumours uses the HUGO Gene Nomenclature Committee (HGNC) system for gene symbols and gene names (https://www.genenames.org/),17 the Human Genome Variation Society (HGVS) recommendations for sequence variants (http://varnomen.hgvs.org/),18 and the reporting guidelines for chromosomal alterations of the International System for Human Cytogenetic Nomenclature 2020.19 Gene symbols are presented in italics, but proteins and gene groups (eg, the family of IDH genes) are not italicized.
A sequence alteration relative to a transcript reference sequence is reported using a “c.” prefix for the coding DNA sequence, followed by the nucleotide number and nucleotide change. The predicted protein sequence change then follows a “p.” prefix with the reference amino acid, the amino acid number, and the variant amino acid resulting from the mutation. For example, the most common BRAF variant is BRAF:c.1799T>A p.Val600Glu (or BRAF:c.1799T>A p.V600E if single-letter amino acid codes are preferred). Notably, this example assumes that a particular BRAF transcript reference sequence accession and version have previously been defined, eg, NM_004333.5.
For some genes, such as those in the H3 histone group, there is potential for confusion with amino acid numbering. Histone amino acid positions are typically described in the context of the protein sequence lacking the initiating methionine, resulting in a single amino acid difference in numbering compared with the predicted sequence derived from the corresponding gene transcript. The description of histone sequence alterations in many cancers has therefore differed to date from the HGVS numbering by omitting the first amino acid. Next-generation sequencing reports, however, follow HGVS guidelines. The coexistence of these 2 nomenclatures may lead to confusion for pathologists, oncologists, and researchers. To address this issue, the fifth edition uses the legacy protein numbering system in parentheses after the protein-level variant description, eg, H3-3A:c.103G>A p.Gly35Arg (G34R), or H3-3A:c.83A>T p.Lys28Met (K27M). In these examples, as noted above, prior definition of the accession and version of the reference transcript is required.
CNS Tumor Grading
CNS tumor grading has for many decades differed from the grading of other, non-CNS neoplasms, since brain and spinal cord tumors have had grades applied across different entities.20 As discussed below, WHO CNS5 has moved CNS tumor grading closer to how grading is done for non-CNS neoplasms but has retained some key aspects of traditional CNS tumor grading because of how embedded such grading has been in neuro-oncology practice. Two specific aspects of CNS tumor grading have changed for WHO CNS5: Arabic numerals are employed (rather than Roman numerals) and neoplasms are graded within types (rather than across different tumor types).14 Nonetheless, because CNS tumor grading still differs from other tumor grading systems, WHO CNS5 endorses use of the term “CNS WHO grade” when assigning grade (eg, see Tables 3–6).
CNS WHO Grades of Selected Types, Covering Entities for Which There Is a New Approach to Grading, an Updated Grade, or a Newly Recognized Tumor That Has an Accepted Grade
| CNS WHO Grades of Selected Types | |
|---|---|
| Astrocytoma, IDH-mutant | 2, 3, 4 |
| Oligodendroglioma, IDH-mutant, and 1p/19q-codeleted | 2, 3 |
| Glioblastoma, IDH-wildtype | 4 |
| Diffuse astrocytoma, MYB- or MYBL1-altered | 1 |
| Polymorphous low-grade neuroepithelial tumor of the young | 1 |
| Diffuse hemispheric glioma, H3 G34-mutant | 4 |
| Pleomorphic xanthoastrocytoma | 2, 3 |
| Multinodular and vacuolating neuronal tumor | 1 |
| Supratentorial ependymomaa | 2, 3 |
| Posterior fossa ependymomaa | 2, 3 |
| Myxopapillary ependymoma | 2 |
| Meningioma | 1, 2, 3 |
| Solitary fibrous tumor | 1, 2, 3 |
| CNS WHO Grades of Selected Types | |
|---|---|
| Astrocytoma, IDH-mutant | 2, 3, 4 |
| Oligodendroglioma, IDH-mutant, and 1p/19q-codeleted | 2, 3 |
| Glioblastoma, IDH-wildtype | 4 |
| Diffuse astrocytoma, MYB- or MYBL1-altered | 1 |
| Polymorphous low-grade neuroepithelial tumor of the young | 1 |
| Diffuse hemispheric glioma, H3 G34-mutant | 4 |
| Pleomorphic xanthoastrocytoma | 2, 3 |
| Multinodular and vacuolating neuronal tumor | 1 |
| Supratentorial ependymomaa | 2, 3 |
| Posterior fossa ependymomaa | 2, 3 |
| Myxopapillary ependymoma | 2 |
| Meningioma | 1, 2, 3 |
| Solitary fibrous tumor | 1, 2, 3 |
Grade is based on natural history and for some tumor types, definite grading criteria and understanding of natural history are not yet known. Note the use of Arabic numerals.
aFor morphologically defined ependymomas.
CNS WHO Grades of Selected Types, Covering Entities for Which There Is a New Approach to Grading, an Updated Grade, or a Newly Recognized Tumor That Has an Accepted Grade
| CNS WHO Grades of Selected Types | |
|---|---|
| Astrocytoma, IDH-mutant | 2, 3, 4 |
| Oligodendroglioma, IDH-mutant, and 1p/19q-codeleted | 2, 3 |
| Glioblastoma, IDH-wildtype | 4 |
| Diffuse astrocytoma, MYB- or MYBL1-altered | 1 |
| Polymorphous low-grade neuroepithelial tumor of the young | 1 |
| Diffuse hemispheric glioma, H3 G34-mutant | 4 |
| Pleomorphic xanthoastrocytoma | 2, 3 |
| Multinodular and vacuolating neuronal tumor | 1 |
| Supratentorial ependymomaa | 2, 3 |
| Posterior fossa ependymomaa | 2, 3 |
| Myxopapillary ependymoma | 2 |
| Meningioma | 1, 2, 3 |
| Solitary fibrous tumor | 1, 2, 3 |
| CNS WHO Grades of Selected Types | |
|---|---|
| Astrocytoma, IDH-mutant | 2, 3, 4 |
| Oligodendroglioma, IDH-mutant, and 1p/19q-codeleted | 2, 3 |
| Glioblastoma, IDH-wildtype | 4 |
| Diffuse astrocytoma, MYB- or MYBL1-altered | 1 |
| Polymorphous low-grade neuroepithelial tumor of the young | 1 |
| Diffuse hemispheric glioma, H3 G34-mutant | 4 |
| Pleomorphic xanthoastrocytoma | 2, 3 |
| Multinodular and vacuolating neuronal tumor | 1 |
| Supratentorial ependymomaa | 2, 3 |
| Posterior fossa ependymomaa | 2, 3 |
| Myxopapillary ependymoma | 2 |
| Meningioma | 1, 2, 3 |
| Solitary fibrous tumor | 1, 2, 3 |
Grade is based on natural history and for some tumor types, definite grading criteria and understanding of natural history are not yet known. Note the use of Arabic numerals.
aFor morphologically defined ependymomas.
Layered Report Structure
| Integrated diagnosis (combined tissue-based histological and molecular diagnosis) |
| Histological diagnosis |
| CNS WHO grade |
| Molecular information (listed) |
| Integrated diagnosis (combined tissue-based histological and molecular diagnosis) |
| Histological diagnosis |
| CNS WHO grade |
| Molecular information (listed) |
Layered Report Structure
| Integrated diagnosis (combined tissue-based histological and molecular diagnosis) |
| Histological diagnosis |
| CNS WHO grade |
| Molecular information (listed) |
| Integrated diagnosis (combined tissue-based histological and molecular diagnosis) |
| Histological diagnosis |
| CNS WHO grade |
| Molecular information (listed) |
Layered Report Example Illustrating: (1) Use of Site in the Diagnosis; (2) Use of a Histological Diagnosis That Does Not Designate “Anaplasia” But the Report Still Assigns a Grade; (3) Use of the NOS Designation (Here Because the Case Could Not Be Worked up Adequately at a Molecular Level)
| Cerebrum | |
|---|---|
| Integrated diagnosis | Supratentorial ependymoma, NOS |
| Histopathological classification | Ependymoma |
| CNS WHO grade | 3 |
| Molecular information | Derivatives extracted from FFPE tissue were of insufficient quality for sequencing and insufficient tissue remained for FISH studies |
| Cerebrum | |
|---|---|
| Integrated diagnosis | Supratentorial ependymoma, NOS |
| Histopathological classification | Ependymoma |
| CNS WHO grade | 3 |
| Molecular information | Derivatives extracted from FFPE tissue were of insufficient quality for sequencing and insufficient tissue remained for FISH studies |
Abbreviations: CNS, central nervous system; FFPE, formalin-fixed paraffin-embedded; FISH, fluorescence in situ hybridization; NOS, not otherwise specified.
Layered Report Example Illustrating: (1) Use of Site in the Diagnosis; (2) Use of a Histological Diagnosis That Does Not Designate “Anaplasia” But the Report Still Assigns a Grade; (3) Use of the NOS Designation (Here Because the Case Could Not Be Worked up Adequately at a Molecular Level)
| Cerebrum | |
|---|---|
| Integrated diagnosis | Supratentorial ependymoma, NOS |
| Histopathological classification | Ependymoma |
| CNS WHO grade | 3 |
| Molecular information | Derivatives extracted from FFPE tissue were of insufficient quality for sequencing and insufficient tissue remained for FISH studies |
| Cerebrum | |
|---|---|
| Integrated diagnosis | Supratentorial ependymoma, NOS |
| Histopathological classification | Ependymoma |
| CNS WHO grade | 3 |
| Molecular information | Derivatives extracted from FFPE tissue were of insufficient quality for sequencing and insufficient tissue remained for FISH studies |
Abbreviations: CNS, central nervous system; FFPE, formalin-fixed paraffin-embedded; FISH, fluorescence in situ hybridization; NOS, not otherwise specified.
Layered Report Example Illustrating: (1) A Tumor Type With a Subtype; (2) Lack of a Definite Grade; and (3) That the Integrated Diagnosis Does Not Necessarily Have the Histological Designation Included
| Cerebrum | |
|---|---|
| Integrated diagnosis | Diffuse low-grade glioma, MAPK pathway-altered Subtype: Diffuse low-grade glioma, FGFR1 TKD-duplicated |
| Histopathological classification | Oligodendroglioma |
| CNS WHO grade | Not assigned |
| Molecular information | Duplication of the FGFR1 tyrosine kinase domain (next-generation sequencing) |
| Cerebrum | |
|---|---|
| Integrated diagnosis | Diffuse low-grade glioma, MAPK pathway-altered Subtype: Diffuse low-grade glioma, FGFR1 TKD-duplicated |
| Histopathological classification | Oligodendroglioma |
| CNS WHO grade | Not assigned |
| Molecular information | Duplication of the FGFR1 tyrosine kinase domain (next-generation sequencing) |
Layered Report Example Illustrating: (1) A Tumor Type With a Subtype; (2) Lack of a Definite Grade; and (3) That the Integrated Diagnosis Does Not Necessarily Have the Histological Designation Included
| Cerebrum | |
|---|---|
| Integrated diagnosis | Diffuse low-grade glioma, MAPK pathway-altered Subtype: Diffuse low-grade glioma, FGFR1 TKD-duplicated |
| Histopathological classification | Oligodendroglioma |
| CNS WHO grade | Not assigned |
| Molecular information | Duplication of the FGFR1 tyrosine kinase domain (next-generation sequencing) |
| Cerebrum | |
|---|---|
| Integrated diagnosis | Diffuse low-grade glioma, MAPK pathway-altered Subtype: Diffuse low-grade glioma, FGFR1 TKD-duplicated |
| Histopathological classification | Oligodendroglioma |
| CNS WHO grade | Not assigned |
| Molecular information | Duplication of the FGFR1 tyrosine kinase domain (next-generation sequencing) |
Arabic vs Roman numerals.
—Traditionally, CNS WHO tumor grades were written as Roman numerals. However, the fifth-edition WHO Blue Books have emphasized more uniform approaches to tumor classification and grading and have favored the use of Arabic numerals for grading, as is currently done for all the other organ systems. Furthermore, a danger of using Roman numerals in a within-tumor grading system is that a “II” and a “III” or a “III” and a “IV” can be mistaken for one another and an uncaught typographical error could have clinical consequences. This was less likely when each tumor type had a different name, eg, “anaplastic” was present in addition to grade “III.” Given these considerations, WHO CNS5 has changed all CNS WHO tumor grades to Arabic numerals (Table 3).
Grading within types.
—As outlined above, CNS tumors have traditionally had a grade assigned to each entity, and grades were applied across different entities.20 For example, in prior WHO classifications, if a tumor had been classified as an anaplastic astrocytoma, it was automatically assigned to WHO grade III (Roman numerals were used for CNS tumor grading in past classifications); there was no option to grade an anaplastic astrocytoma as WHO grade I, II, or IV. Notably, an anaplastic (malignant) meningioma was also assigned to WHO grade III. Even though tumors like meningiomas and astrocytomas are biologically unrelated, WHO grade III tumors in these different categories were expected to have roughly similar survival times. But these were only roughly similar, with the clinical course of an anaplastic astrocytoma often quite different from that of an anaplastic (malignant) meningioma. This approach thus correlated grade to an idealized clinical-biological behavior; for instance, WHO grade I tumors were curable if they could be surgically removed; at the other end of the spectrum, WHO grade IV tumors were highly malignant, leading to death in relatively short periods of time in the absence of effective therapy.
This entity-specific and clinical approach to tumor grading was different from the grading used in other, non-CNS tumor types.20 Most tumors in other organ systems are graded within tumor types, eg, a breast or prostate cancer is graded according to its particular grading system. In the 2016 CNS WHO classification, solitary fibrous tumor/ hemangiopericytoma was graded in this manner, using a single name but with the option of 3 grades. In WHO CNS5, the shift to within-tumor-type grading has been extended to many categories (eg, see Tables 3 and 5). This change was done for several reasons: (1) to provide more flexibility in using grade relative to the tumor type, (2) to emphasize biological similarities within tumor types rather than approximate clinical behavior, and (3) to conform with WHO grading in non-CNS tumor types.
“Clinicopathological” grading.
—Nonetheless, because CNS tumor grading has for decades been linked to overall expected clinical-biological behaviors (see above), WHO CNS5 has generally retained the ranges of grades used for tumor types in prior editions. In this context, IDH-mutant astrocytomas extend from CNS WHO grade 2-4 and meningiomas from CNS WHO grade 1-3. In other words, at least for now, there is neither a CNS WHO grade 1 IDH-mutant astrocytoma nor a CNS WHO grade 4 meningioma. Moreover, given that tumors are graded on the basis of their expected natural history, certain malignant tumors (eg, medulloblastoma, germinoma) can be assigned a CNS WHO grade 4 designation in WHO CNS5 even if they now have effective treatments associated with favorable survival times, particularly in the case of certain molecularly defined types like WNT-activated medulloblastoma.
The above approach to grading is a compromise since the original underlying prognostic correlations were based on natural history, at a time when few effective therapies were available. Today, estimating natural history is nearly impossible, since practically all patients receive therapies that often affect overall survival.21 In the context of modern therapies that can dramatically affect patient survival, the necessity of grading every tumor type is questionable. In fact, in editorial discussions for WHO CNS5, it was argued that grades should not be assigned if designation of a grade could confuse clinical care (eg, see Table 6). For instance, WNT-activated medulloblastoma is an embryonal tumor that has an aggressive behavior if left untreated but that is responsive to current therapeutic regimens such that nearly all patients have long-term survival. Designating this tumor as CNS WHO grade 4, and therefore equivalent to many untreatable pediatric brain tumors with a dismal outcome, potentially risks giving a false sense of prognosis when therapeutic options are discussed in the clinic. Conversely, designating this tumor as CNS WHO grade 1 on the basis of its good outcome, and therefore equivalent to neoplasms with a similar prognosis on the basis of surgery alone, certainly gives a false sense that the tumor is biologically benign.
Combined histological and molecular grading.
—Traditionally, CNS tumor grading has been based exclusively on histological features, but certain molecular markers can now provide powerful prognostic information. For this reason, molecular parameters have now been added as biomarkers of grading and for further estimating prognosis within multiple tumor types. Examples in WHO CNS5 include CDKN2A/B homozygous deletion in IDH-mutant astrocytomas, as well as TERT promoter mutation, EGFR amplification, and +7/−10 copy number changes in IDH-wildtype diffuse astrocytomas (allowing a glioblastoma, IDH-wildtype CNS WHO grade 4 designation even in cases that otherwise appear histologically lower grade). In other words, a molecular parameter can sometimes add value to histological findings in assigning a grade. Specific instances are discussed for the relevant tumor types (see below). It is also important to note that CNS WHO grade is therefore no longer restricted to being a histological grade, as was previously recommended.22
NOS (Not Otherwise Specified) and NEC (Not Elsewhere Classified) Diagnoses
As detailed elsewhere,12,13 use of the suffixes NOS and NEC allow the ready separation of standard, well-characterized WHO diagnoses from those diagnoses that result from either (1) a lack of necessary diagnostic (eg, molecular) information or (2) nondiagnostic (ie, for a WHO diagnosis) or negative results. Adding an NOS suffix indicates that the diagnostic information (histological or molecular) necessary to assign a specific WHO diagnosis is not available, providing an alert to the oncologist that a molecular work-up has not been undertaken or failed technically. An NEC suffix, on the other hand, indicates that the necessary diagnostic testing has been successfully performed but that the results do not readily allow for a WHO diagnosis; for example, if there is a mismatch between clinical, histological, immunohistochemical, and/or genetic features. NEC diagnoses are what pathologists have termed “descriptive diagnoses,” in which the pathologist uses a non-WHO diagnosis to categorize the tumor. In this regard, an NEC designation provides an alert to the oncologist that, despite an adequate pathological work-up, the tumor does not conform to a standard WHO diagnosis. Like WHO diagnoses, NEC and NOS diagnoses are facilitated by the use of layered integrated reports22 (see below and Tables 4-6).
Novel Diagnostic Technologies
Over the past century, many novel technologies have impacted tumor classification. These have included light microscopy, histochemical stains, electron microscopy, immunohistochemistry, molecular genetics, and most recently, a variety of broad molecular profiling approaches. Each burst on the scene as a method that promised to change classification completely and each then eventually found a specific niche alongside the others, rather than replacing them. Over the past couple of decades, nucleic acid-based methodologies (eg, DNA and RNA sequencing, DNA fluorescence in situ hybridization, RNA expression profiling) have clearly shown their abilities to contribute to tumor diagnosis and classification, as evidenced by the changes in the updated fourth edition (2016) and in WHO CNS5. The availability of such technologies was increasing throughout the world as the 2016 classification was being prepared,23,24 and the last few years have witnessed further expansion of availability as well as skillful ways to adapt to molecular classification recommendations.25,26 WHO CNS5 thus incorporates more molecular approaches for the classification of CNS tumors.
Over the past decade, methylome profiling—the use of arrays to determine DNA methylation patterns across the genome—has emerged as a powerful approach to CNS tumor classification, as detailed in a variety of publications over the past few years.27–30 Most CNS tumor types can be reliably identified by their methylome profile, although caveats remain that optimal methodologic approaches and regulatory issues for methylome profiling have yet to be resolved and that the technology is currently not widely available.14 Copy number profiles can also be derived from methylation data, eg, 1p/19q codeletion, the +7/−10 signature, amplifications, homozygous deletions, and profiles suggestive of fusion events. At this time, methylome profiling is an effective ancillary method for brain and spinal cord tumor classification when used alongside other, standard technologies, including histology. Indeed, the great majority of tumor types and subtypes can also be reliably identified by other techniques, eg, from a combination of morphological features and defining genetic alteration. On the other hand, methylome profiling may be the most effective way to characterize some tumors with unusual morphological features and may be the only current way to identify some rare tumor types and subtypes. The method also has utility when small biopsy samples are limiting for standard technologies. Methylome profiling may also be used as a surrogate marker for genetic events, for instance when a methylome signature is characteristic of an IDH-wildtype glioblastoma in the absence of IDH mutation testing—but methylome profiling cannot serve as a surrogate when targeted therapies and clinical trials require the demonstration of specific mutations prior to patient treatment. For methylome profiling results, careful attention must be paid to the common calibrated score threshold; as discussed in detail elsewhere,28 thresholds may be set at 0.84 or 0.90, and pathologists should be wary about endorsing suggested diagnoses with scores below 0.84 and should probably discard recommendations if scores are below 0.50. As with other diagnostic tests, the pathologist must take into account histological features (eg, tumor cell amount and purity) when interpreting results; for example, methylome profiling can struggle with classification of low-grade diffuse gliomas. For the WHO CNS5, therefore, it is assumed that nearly all (but not all) tumor types are aligned to a distinct methylation signature27 and these are not specified in every Definition; however, information about diagnostic methylation profiling is included in those Definitions and Essential and Desirable Diagnostic Criteria sections for which the method can provide more critical guidance for diagnosis.
Integrated and Layered Diagnoses
Because of the growing importance of molecular information in CNS tumor classification, diagnoses and diagnostic reports need to combine different data types into a single, “integrated” diagnosis. Such integrated diagnoses are implicit in the use of WHO CNS5. Even diagnostic terms that do not incorporate a molecular term may require a molecular characteristic for diagnosis (eg, AT/RT). Thus, to display the full range of diagnostic information available, the use of layered (or tiered) diagnostic reports is strongly encouraged, as endorsed by the International Society of Neuropathology—Haarlem consensus guidelines22 and the International Collaboration on Cancer Reporting.31 Such reports feature an integrated diagnosis at the top, followed by layers that display histological, molecular, and other key types of information (Table 4).
For some tumor types in WHO CNS5, the listed diagnostic terms are general ones (eg, Diffuse high-grade pediatric-type glioma, H3-wildtype and IDH-wildtype and Diffuse low-grade glioma, MAPK pathway-altered); for these types, a combination of diagnostic features drawn from a matrix of relevant histological and molecular abnormalities is necessary to arrive at a specific integrated diagnosis. These approaches are described for each of these tumor groups and are similar to how the 2016 CNS WHO classified medulloblastomas22 and what cIMPACT-NOW Update 4 recommended for pediatric low-grade diffuse gliomas10: an integrated diagnosis optimally combines a term from a histologically defined list of tumors and a genetically defined list of tumors (Tables 4–6). Even though each list may contain many items, some combinations are more common than others. The resulting number of routinely used integrated diagnoses is typically manageable, and common diagnoses are included as tumor subtypes in the case of Diffuse low-grade glioma, MAPK pathway-altered.
In WHO CNS5, Essential and Desirable Diagnostic Criteria are given for each tumor type, mostly in tabular form, in the hope that such a format makes it easier for the user to evaluate whether key diagnostic criteria are present and whether the combinations of such criteria are sufficient for diagnosis. Essential Diagnostic Criteria are considered “must have” features, but there may be different combinations that allow a diagnosis, ie, not all criteria are needed for a diagnosis. For these diagnostic types, the user should pay close attention to the use of “AND” vs “OR” designations in the Essential Diagnostic Criteria tables. On the other hand, Desirable Diagnostic Criteria are “nice to have” features, ie, they clearly support a diagnosis but are not needed per se.
Newly Recognized Entities and Revised Nomenclature
The major specific changes to the classification are discussed in sections relating to families of tumors below. Multiple newly recognized types (see Table 7) have been accepted into WHO CNS5, and some of the more distinct microscopic features are illustrated in Figures 1–8. In addition, changes were made to the nomenclature of some entities, both to clarify molecular alterations and to follow the nomenclature guidelines in cIMPACT-NOW Update 614 (see Table 8). Other nomenclature changes were made to standardize type names with those in other Blue Books, eg, for peripheral nerve and other soft tissue tumors.
Newly Recognized Tumor Types in the 2021 WHO Classification of Tumors of the Central Nervous System
| Newly Recognized Tumor Types |
|---|
| Diffuse astrocytoma, MYB- or MYBL1-altered |
| Polymorphous low-grade neuroepithelial tumor of the young |
| Diffuse low-grade glioma, MAPK pathway-altered |
| Diffuse hemispheric glioma, H3 G34-mutant |
| Diffuse pediatric-type high-grade glioma, H3-wildtype and IDH-wildtype |
| Infant-type hemispheric glioma |
| High-grade astrocytoma with piloid features |
| Diffuse glioneuronal tumor with oligodendroglioma-like features and nuclear clusters (provisional type) |
| Myxoid glioneuronal tumor |
| Multinodular and vacuolating neuronal tumor |
| Supratentorial ependymoma, YAP1 fusion-positive |
| Posterior fossa ependymoma, group PFA |
| Posterior fossa ependymoma, group PFB |
| Spinal ependymoma, MYCN-amplified |
| Cribriform neuroepithelial tumor (provisional type) |
| CNS neuroblastoma, FOXR2-activated |
| CNS tumor with BCOR internal tandem duplication |
| Desmoplastic myxoid tumor of the pineal region, SMARCB1-mutant |
| Intracranial mesenchymal tumor, FET-CREB fusion positive (provisional type) |
| CIC-rearranged sarcoma |
| Primary intracranial sarcoma, DICER1-mutant |
| Pituitary blastoma |
| Newly Recognized Tumor Types |
|---|
| Diffuse astrocytoma, MYB- or MYBL1-altered |
| Polymorphous low-grade neuroepithelial tumor of the young |
| Diffuse low-grade glioma, MAPK pathway-altered |
| Diffuse hemispheric glioma, H3 G34-mutant |
| Diffuse pediatric-type high-grade glioma, H3-wildtype and IDH-wildtype |
| Infant-type hemispheric glioma |
| High-grade astrocytoma with piloid features |
| Diffuse glioneuronal tumor with oligodendroglioma-like features and nuclear clusters (provisional type) |
| Myxoid glioneuronal tumor |
| Multinodular and vacuolating neuronal tumor |
| Supratentorial ependymoma, YAP1 fusion-positive |
| Posterior fossa ependymoma, group PFA |
| Posterior fossa ependymoma, group PFB |
| Spinal ependymoma, MYCN-amplified |
| Cribriform neuroepithelial tumor (provisional type) |
| CNS neuroblastoma, FOXR2-activated |
| CNS tumor with BCOR internal tandem duplication |
| Desmoplastic myxoid tumor of the pineal region, SMARCB1-mutant |
| Intracranial mesenchymal tumor, FET-CREB fusion positive (provisional type) |
| CIC-rearranged sarcoma |
| Primary intracranial sarcoma, DICER1-mutant |
| Pituitary blastoma |
Newly Recognized Tumor Types in the 2021 WHO Classification of Tumors of the Central Nervous System
| Newly Recognized Tumor Types |
|---|
| Diffuse astrocytoma, MYB- or MYBL1-altered |
| Polymorphous low-grade neuroepithelial tumor of the young |
| Diffuse low-grade glioma, MAPK pathway-altered |
| Diffuse hemispheric glioma, H3 G34-mutant |
| Diffuse pediatric-type high-grade glioma, H3-wildtype and IDH-wildtype |
| Infant-type hemispheric glioma |
| High-grade astrocytoma with piloid features |
| Diffuse glioneuronal tumor with oligodendroglioma-like features and nuclear clusters (provisional type) |
| Myxoid glioneuronal tumor |
| Multinodular and vacuolating neuronal tumor |
| Supratentorial ependymoma, YAP1 fusion-positive |
| Posterior fossa ependymoma, group PFA |
| Posterior fossa ependymoma, group PFB |
| Spinal ependymoma, MYCN-amplified |
| Cribriform neuroepithelial tumor (provisional type) |
| CNS neuroblastoma, FOXR2-activated |
| CNS tumor with BCOR internal tandem duplication |
| Desmoplastic myxoid tumor of the pineal region, SMARCB1-mutant |
| Intracranial mesenchymal tumor, FET-CREB fusion positive (provisional type) |
| CIC-rearranged sarcoma |
| Primary intracranial sarcoma, DICER1-mutant |
| Pituitary blastoma |
| Newly Recognized Tumor Types |
|---|
| Diffuse astrocytoma, MYB- or MYBL1-altered |
| Polymorphous low-grade neuroepithelial tumor of the young |
| Diffuse low-grade glioma, MAPK pathway-altered |
| Diffuse hemispheric glioma, H3 G34-mutant |
| Diffuse pediatric-type high-grade glioma, H3-wildtype and IDH-wildtype |
| Infant-type hemispheric glioma |
| High-grade astrocytoma with piloid features |
| Diffuse glioneuronal tumor with oligodendroglioma-like features and nuclear clusters (provisional type) |
| Myxoid glioneuronal tumor |
| Multinodular and vacuolating neuronal tumor |
| Supratentorial ependymoma, YAP1 fusion-positive |
| Posterior fossa ependymoma, group PFA |
| Posterior fossa ependymoma, group PFB |
| Spinal ependymoma, MYCN-amplified |
| Cribriform neuroepithelial tumor (provisional type) |
| CNS neuroblastoma, FOXR2-activated |
| CNS tumor with BCOR internal tandem duplication |
| Desmoplastic myxoid tumor of the pineal region, SMARCB1-mutant |
| Intracranial mesenchymal tumor, FET-CREB fusion positive (provisional type) |
| CIC-rearranged sarcoma |
| Primary intracranial sarcoma, DICER1-mutant |
| Pituitary blastoma |
Tumor Types With Revised Nomenclature or Revised Placement in the 2021 WHO Classification of Tumors of the Central Nervous System
| Tumor Types With Revised Nomenclature or Revised Placement |
|---|
| Astrocytoma, IDH-mutant (covers grades 2-4; eliminates the term “Glioblastoma, IDH-mutant”) |
| Diffuse midline glioma, H3 K27-altered (changes “mutant” to “altered” given multiple mechanisms) |
| Chordoid glioma (removes site designation) |
| Astroblastoma, MN1-altered (adds genetic modifier) |
| Supratentorial ependymoma, ZFTA fusion-positive (reflects changes in fusion partner and gene nomenclature; see text) |
| Embryonal tumor with multilayered rosettes (removes genetic modifier to allow for genetic subtypes) |
| Malignant melanotic nerve sheath tumor (conforms to terminology in soft tissue pathology literature) |
| Solitary fibrous tumor (removes the term “hemangiopericytoma” to conform fully with soft tissue pathology nomenclature) |
| Mesenchymal chondrosarcoma (formerly a subtype) |
| Adamantinomatous craniopharyngioma (formerly a subtype) |
| Papillary craniopharyngioma (formerly a subtype) |
| Pituicytoma, granular cell tumor of the sellar region, and spindle cell oncocytoma (grouped rather than separate) |
| Pituitary adenoma/PitNET (adds the term “PitNET”) |
| Tumor Types With Revised Nomenclature or Revised Placement |
|---|
| Astrocytoma, IDH-mutant (covers grades 2-4; eliminates the term “Glioblastoma, IDH-mutant”) |
| Diffuse midline glioma, H3 K27-altered (changes “mutant” to “altered” given multiple mechanisms) |
| Chordoid glioma (removes site designation) |
| Astroblastoma, MN1-altered (adds genetic modifier) |
| Supratentorial ependymoma, ZFTA fusion-positive (reflects changes in fusion partner and gene nomenclature; see text) |
| Embryonal tumor with multilayered rosettes (removes genetic modifier to allow for genetic subtypes) |
| Malignant melanotic nerve sheath tumor (conforms to terminology in soft tissue pathology literature) |
| Solitary fibrous tumor (removes the term “hemangiopericytoma” to conform fully with soft tissue pathology nomenclature) |
| Mesenchymal chondrosarcoma (formerly a subtype) |
| Adamantinomatous craniopharyngioma (formerly a subtype) |
| Papillary craniopharyngioma (formerly a subtype) |
| Pituicytoma, granular cell tumor of the sellar region, and spindle cell oncocytoma (grouped rather than separate) |
| Pituitary adenoma/PitNET (adds the term “PitNET”) |
Tumor Types With Revised Nomenclature or Revised Placement in the 2021 WHO Classification of Tumors of the Central Nervous System
| Tumor Types With Revised Nomenclature or Revised Placement |
|---|
| Astrocytoma, IDH-mutant (covers grades 2-4; eliminates the term “Glioblastoma, IDH-mutant”) |
| Diffuse midline glioma, H3 K27-altered (changes “mutant” to “altered” given multiple mechanisms) |
| Chordoid glioma (removes site designation) |
| Astroblastoma, MN1-altered (adds genetic modifier) |
| Supratentorial ependymoma, ZFTA fusion-positive (reflects changes in fusion partner and gene nomenclature; see text) |
| Embryonal tumor with multilayered rosettes (removes genetic modifier to allow for genetic subtypes) |
| Malignant melanotic nerve sheath tumor (conforms to terminology in soft tissue pathology literature) |
| Solitary fibrous tumor (removes the term “hemangiopericytoma” to conform fully with soft tissue pathology nomenclature) |
| Mesenchymal chondrosarcoma (formerly a subtype) |
| Adamantinomatous craniopharyngioma (formerly a subtype) |
| Papillary craniopharyngioma (formerly a subtype) |
| Pituicytoma, granular cell tumor of the sellar region, and spindle cell oncocytoma (grouped rather than separate) |
| Pituitary adenoma/PitNET (adds the term “PitNET”) |
| Tumor Types With Revised Nomenclature or Revised Placement |
|---|
| Astrocytoma, IDH-mutant (covers grades 2-4; eliminates the term “Glioblastoma, IDH-mutant”) |
| Diffuse midline glioma, H3 K27-altered (changes “mutant” to “altered” given multiple mechanisms) |
| Chordoid glioma (removes site designation) |
| Astroblastoma, MN1-altered (adds genetic modifier) |
| Supratentorial ependymoma, ZFTA fusion-positive (reflects changes in fusion partner and gene nomenclature; see text) |
| Embryonal tumor with multilayered rosettes (removes genetic modifier to allow for genetic subtypes) |
| Malignant melanotic nerve sheath tumor (conforms to terminology in soft tissue pathology literature) |
| Solitary fibrous tumor (removes the term “hemangiopericytoma” to conform fully with soft tissue pathology nomenclature) |
| Mesenchymal chondrosarcoma (formerly a subtype) |
| Adamantinomatous craniopharyngioma (formerly a subtype) |
| Papillary craniopharyngioma (formerly a subtype) |
| Pituicytoma, granular cell tumor of the sellar region, and spindle cell oncocytoma (grouped rather than separate) |
| Pituitary adenoma/PitNET (adds the term “PitNET”) |
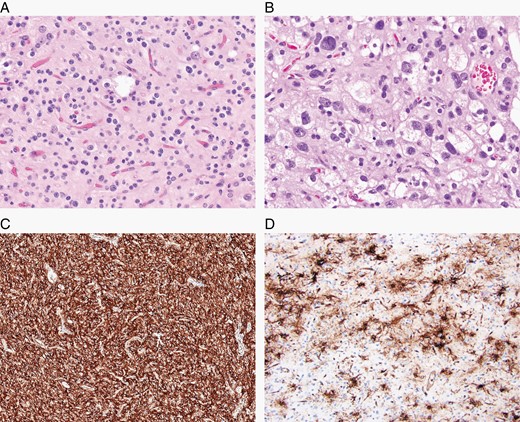
Polymorphous low-grade neuroepithelial tumor of the young (PLNTY) is a glial neoplasm associated with a history of epilepsy in young people, diffuse growth patterns, frequent presence of oligodendroglioma-like components, calcification, CD34 immunoreactivity, and MAPK pathway-activating genetic abnormalities. (A) Common oligodendroglioma-like appearance (H&E, ×200), but (B) histological appearances can vary greatly within tumors (H&E, ×400). (C) CD34 immunostaining is typically strong and diffuse in the tumor (×100); and (D) is often found in the peritumoral cortex (×200).
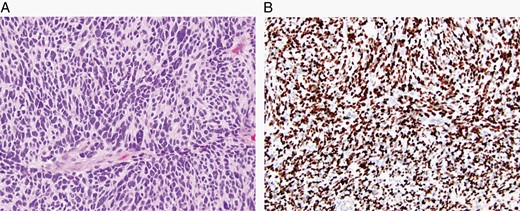
Diffuse hemispheric glioma, H3 G34-mutant, is a malignant, infiltrative glioma, typically of the cerebral hemispheres and with a missense mutation in the H3F3A gene that results in a G34R/V substitution of histone H3. (A) High-grade anaplastic features, sometimes with an embryonal appearance (H&E, ×200) and (B) positive nuclear staining with H3 G34R/V immunohistochemistry (×100).
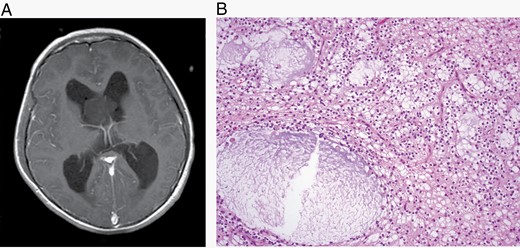
Myxoid glioneuronal tumor is a tumor typically arising in the septal region and involving the lateral ventricle. It is characterized by a proliferation of oligodendrocyte-like tumor cells embedded in a prominent myxoid stroma, often including admixed floating neurons, neurocytic rosettes, and/or perivascular neuropil, and by a dinucleotide mutation in the PDGFRA gene. (A) Common septal location (magnetic resonance imaging, T1 with contrast) and (B) characteristic histological features with small round cells and myxoid stroma (H&E, ×200).
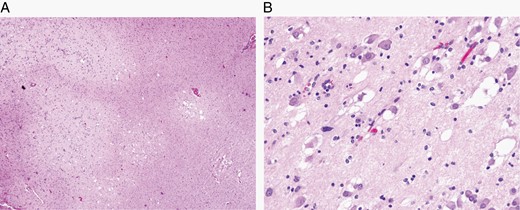
Multinodular and vacuolating neuronal tumor is a benign tumor comprising monomorphous neuronal elements in discrete and coalescent nodules, with vacuolar changes both in tumor cells and the neuropil. (A) Multinodular appearance (H&E, ×40). (B) Vacuolar change in tumor cells and in neuropil (H&E, ×200).
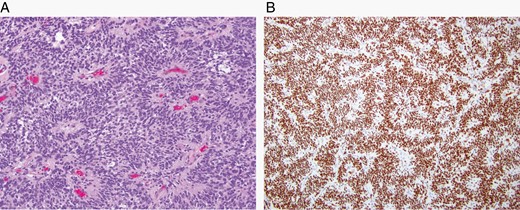
CNS tumor with BCOR internal tandem duplication is a neoplasm with a mostly solid growth pattern, uniform oval or spindle-shaped cells, a dense capillary network, focal pseudorosette formation, and an internal tandem duplication (ITD) in exon 15 of the BCOR gene. (A) High-grade neoplasm with perivascular rosettes (H&E, ×200) and (B) strong, diffuse nuclear staining on BCOR immunohistochemistry (×100).
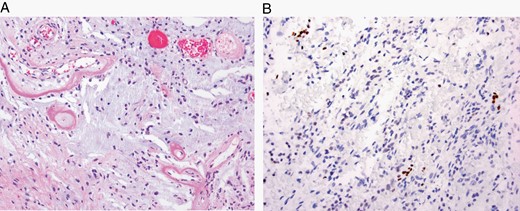
Desmoplastic myxoid tumor of the pineal region, SMARCB1-mutant is a rare pineal-region tumor that features desmoplasia and myxoid changes (H&E, ×200) (A) as well as loss of INI1 staining (×200) (B).
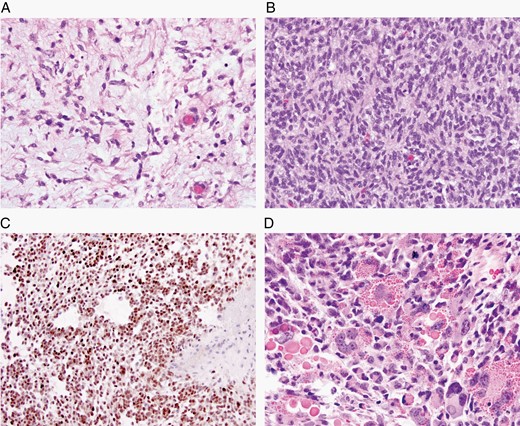
Newly recognized mesenchymal, non-meningothelial tumors of uncertain histogenesis. (A) Intracranial mesenchymal tumor, FET-CREB fusion-positive (H&E, ×200); these tumors have variable morphology and a fusion of an FET RNA-binding protein family gene and a member of the CREB family of transcription factors. (B, C) CIC-rearranged sarcoma, with (B) poorly differentiated cells (H&E, ×200) and (C) with ETV4 frequently being upregulated in these tumors (×200). (D) Primary intracranial sarcoma, DICER1-mutant with characteristic eosinophilic cytoplasmic droplets (H&E, ×200).
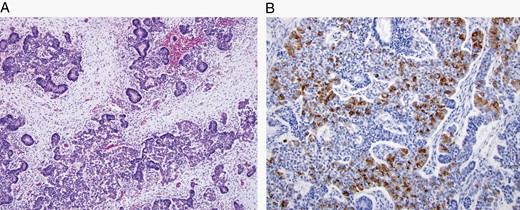
Pituitary blastoma is a malignant embryonal sellar neoplasm composed of primitive blastemal cells, neuroendocrine cells, and Rathke epithelium, typically occurring in young children and linked to germline or somatic variants in the DICER1 gene. (A) Neuroendocrine cells arranged in lobules, rosettes, and glands, interspersed with small undifferentiated, blastemal cells (H&E, ×100) and with (B) ACTH immunoreactivity in some cells (×200).
Some proposed tumor types were discussed and provisionally accepted as types because they appeared to be clinicopathologically distinct, but additional published studies are needed for full acceptance. The 3 provisional entities are designated in italics in the table: Diffuse glioneuronal tumor with oligodendroglioma-like features and nuclear clusters (DGONC); Cribriform neuroepithelial tumor (CRINET); and Intracranial mesenchymal tumor, FET-CREB fusion-positive. Others were discussed during the editorial process, but the published literature still left questions about the nature of the proposed entity. An example of this was Neuroepithelial tumor, PATZ1 fusion-positive, for which there are only a few cases described in the literature.32–34 While unpublished data suggest that these lesions have distinct molecular alterations, there is marked heterogeneity in their histopathological appearances and clinical courses, and therefore more published data are needed to evaluate whether these cases form a distinct tumor type.
Specific Changes
Gliomas, Glioneuronal Tumors, and Neuronal Tumors
WHO CNS5 has taken a new approach to classify the Gliomas, Glioneuronal Tumors, and Neuronal Tumors, and dividing them into 6 different families: (1) Adult-type diffuse gliomas (the majority of primary brain tumors in neuro-oncology practice of adults, eg, glioblastoma, IDH-wildtype); (2) Pediatric-type diffuse low-grade gliomas (expected to have good prognoses); (3) Pediatric-type diffuse high-grade gliomas (expected to behave aggressively); (4) Circumscribed astrocytic gliomas (“circumscribed” referring to their more solid growth pattern, as opposed to the inherently “diffuse” tumors in groups 1, 2, and 3); (5) Glioneuronal and neuronal tumors (a diverse group of tumors, featuring neuronal differentiation); and (6) Ependymomas (now classified by site as well as histological and molecular features). Choroid Plexus Tumors, with their marked epithelial characteristics, are separated from the category of Gliomas, Glioneuronal Tumors, and Neuronal Tumors.
Fourteen newly recognized types have been added to the classification of Gliomas, Glioneuronal Tumors, and Neuronal Tumors (see Table 7). For some of these types—especially for Diffuse high-grade pediatric-type, H3-wildtype and IDH-wildtype, and for Diffuse low-grade glioma, MAPK pathway-altered—integrating histological appearances and molecular features is required to arrive at a diagnosis, and such data are most effectively displayed as tiers of information. There have also been some nomenclature changes to existing entities. For example, the diffuse midline glioma is now designated as “H3 K27-altered” rather than “H3 K27M-mutant” in order to recognize alternative mechanisms by which the pathogenic pathway can be altered in these tumors.35 Astroblastoma has been specified as “MN1-altered” to provide more diagnostic focus for this entity, even though future work will be needed to establish clear histopathological and molecular parameters by which astroblastomas with MN1 alterations can be distinguished from morphologically comparable neuroepithelial tumors with similar genetic alterations. For other tumor types, changes in nomenclature regarding the inclusion of genetic and anatomical site modifiers have followed the recommendations of cIMPACT-NOW Update 614 and cIMPACT-NOW Update 7.16 Nearly all of these newly recognized types can be diagnosed on the basis of standard histological, immunohistochemical, and molecular analyses.
Division of diffuse gliomas into adult-type and pediatric-type.
—Importantly, WHO CNS5 recognizes the clinical and molecular distinctions between those diffuse gliomas that primarily occur in adults (termed “adult-type”) and those that occur primarily in children (termed “pediatric-type”). Note the use of the word “primarily” in the last sentence, since pediatric-type tumors may sometimes occur in adults, particularly young adults, and adult-type tumors may more rarely occur in children. Nonetheless, the division of the classification into adult-type and pediatric-type diffuse gliomas should be a step forward in clearly separating these prognostically and biologically distinct groups of tumors. The need to do so has been considered for a long time, but the elucidation of molecular differences has now made this possible. It is hoped that this distinction will enable improved care for both children and adults with CNS tumors.
Simplification of the classification of common, adult-type, diffuse gliomas.
—In the updated fourth edition CNS classification from 2016, the common diffuse gliomas of adults were divided into 15 entities, largely because different grades were assigned to different entities (eg, Anaplastic oligodendroglioma was considered a different type from Oligodendroglioma) and because NOS designations were assigned to distinct entities (eg, Diffuse astrocytoma, NOS). WHO CNS5, on the other hand, includes only 3 types: Astrocytoma, IDH-mutant; Oligodendroglioma, IDH-mutant and 1p/19q-codeleted; and Glioblastoma, IDH-wildtype.
This focusing of the classification has resulted from (1) more ecumenical use of NOS and NEC terminology, as discussed above and in cIMPACT-NOW Update 112; (2) recognition of the value of molecular diagnostics to assign poorly defined entities (eg, oligoastrocytomas or IDH-wildtype diffuse astrocytic tumors) to more objectively defined types; and (3) use of grades within types14,15 rather than requiring each grade to have a different name (see above). In addition, in the fifth edition WHO Blue Books, subtypes (eg, Gliosarcoma and Giant cell glioblastoma) are not listed in the classification, but these classic variants are discussed in their respective chapters.
Nomenclature and grading of common, adult-type, diffuse astrocytic gliomas.
—In the 2016 WHO classification, IDH-mutant diffuse astrocytic tumors were assigned to 3 different tumor types (Diffuse astrocytoma, Anaplastic astrocytoma, and Glioblastoma) depending on histological parameters. In the current classification, however, all IDH-mutant diffuse astrocytic tumors are considered a single type (Astrocytoma, IDH-mutant) and are then graded as CNS WHO grade 2, 3, or 4. Moreover, grading is no longer entirely histological, since the presence of CDKN2A/B homozygous deletion results in a CNS WHO grade of 4, even in the absence of microvascular proliferation or necrosis.
For IDH-wildtype diffuse astrocytic (NB: diffuse and astrocytic) tumors in adults, a number of papers have shown that the presence of 1 or more of 3 genetic parameters (TERT promoter mutation, EGFR gene amplification, combined gain of entire chromosome 7 and loss of entire chromosome 10 [+7/−10]) appears sufficient to assign the highest WHO grade.11,36 WHO CNS5 therefore incorporates these 3 genetic parameters as criteria for a diagnosis of Glioblastoma, IDH-wildtype. As a result, Glioblastoma, IDH-wildtype should be diagnosed in the setting of an IDH-wildtype diffuse and astrocytic glioma in adults if there is microvascular proliferation or necrosis or TERT promoter mutation or EGFR gene amplification or +7/−10 chromosome copy number changes. In IDH-wildtype diffuse astrocytomas occurring in younger age groups, however, consideration should be given to the different types of diffuse pediatric-type gliomas (see below).
Pediatric-type low-grade and high-grade diffuse gliomas.
—Two new families of tumor types have been added to the classification to reflect the practical and conceptual importance of separating pediatric-type gliomas from other diffuse gliomas: one for Pediatric-type diffuse low-grade gliomas and one for Pediatric-type diffuse high-grade gliomas. The low-grade group includes 4 entities that feature diffuse growth in the brain but with sometimes overlapping and less specific histological features; in all, molecular work-up helps to characterize the lesion as one type or the other. For CNS5, the 4 types are Diffuse astrocytoma, MYB- or MYBL1-altered; Angiocentric glioma; Polymorphous low-grade neuroepithelial tumor of the young (often abbreviated as PLNTY; Figure 1); and Diffuse low-grade glioma, MAPK pathway-altered. The last of these diagnoses encompass tumors with an astrocytic or oligodendroglial morphology. For these tumors (as for most other glioma types), precise classification requires molecular characterization and the integration of histopathological and molecular information in a tiered diagnostic format.22 Clear delineation of the specific molecular features, in turn, sets the stage for targeted therapies of such tumors.
The high-grade family also comprises 4 types: Diffuse midline glioma, H3 K27-altered; Diffuse hemispheric glioma, H3 G34-mutant (Figure 2); Diffuse pediatric-type high-grade glioma, H3-wildtype and IDH-wildtype; and Infant-type hemispheric glioma. Diffuse midline glioma, H3 K27-altered had been in the 2016 classification, but as mentioned above, its name has been changed to reflect the fact that other changes (eg, EZHIP protein overexpression) can define this entity in addition to the previously recognized H3 K27 mutations. The other 3 are newly recognized types. Diffuse pediatric-type high-grade glioma, H3-wildtype and IDH-wildtype is specified as being wildtype for both H3 and IDH gene families and, like many other CNS tumor types, requires molecular characterization and integration of histopathological and molecular data for diagnostic purposes. Infant-type hemispheric glioma is a novel type of high-grade glioma that occurs in newborns and infants and that has a distinct molecular profile, with fusion genes involving ALK, ROS1, NTRK1/2/3, or MET.37,38 Of note, the term “glioblastoma” is no longer used in the setting of a pediatric-type neoplasm.
Neuronal and glioneuronal tumors.
—All tumors with a neuronal component have remained grouped together in WHO CNS5. Three new types have been added, although the first is provisional (ie, will likely become a fully recognized type in a future classification but currently awaits further published characterizations): DGONC (provisional); Myxoid glioneuronal tumor (Figure 3); and Multinodular and vacuolating neuronal tumor (Figure 4), which had been discussed in the 2016 classification in the chapter on Gangliocytoma.
Ependymomas.
—Ependymomas should now be classified according to a combination of histopathological and molecular features as well as anatomic site,16 thus dividing them into molecular groups across the supratentorial, posterior fossa (PF), and spinal compartments (Table 1).39 WHO CNS5 also now lists 2 molecularly defined types of supratentorial ependymoma: one with ZFTA (the new designation for C11orf95, which is considered more representative of the tumor type than RELA because it may be fused with partners more than RELA) fusion and another with YAP1 fusion. It also now includes 2 molecularly defined types of PF ependymoma, group PFA and group PFB, as well as a spinal tumor defined by the presence of MYCN amplification. Also listed are ependymomas defined by anatomic location but not by a molecular alteration; these can be used either when molecular analysis finds a different molecular alteration to one used to define ependymomas at a particular site or when molecular analysis fails or is unavailable. As described above, the former situation utilizes the NEC suffix and the latter utilizes the NOS suffix. Myxopapillary ependymoma and Subependymoma remain tumor types; currently, although these can be identified with methylome studies, molecular classification does not provide added clinicopathological utility for these 2 tumors.16 In contrast to previous WHO classifications, the myxopapillary ependymoma is now considered CNS WHO grade 2 rather than 1, since its likelihood of recurrence is now understood to be similar to conventional spinal ependymoma. Papillary, clear cell, and tanycytic morphological variants are no longer listed as subtypes of ependymoma, being included instead as patterns in the histopathological description of ependymoma.
Longstanding controversy surrounds the reproducibility and clinicopathological utility of grading ependymal tumors,40 although use of WHO grade in the therapeutic stratification of adult patients with supratentorial ependymoma remains established practice41 while the full clinical associations of molecular alterations in this patient population are being evaluated. WHO CNS5 allows only a histologically defined diagnosis of Ependymoma to be made at any of the 3 anatomic sites; the term “anaplastic ependymoma” is no longer listed.16 Nonetheless, as for other tumors in WHO CNS5, a pathologist can still choose to assign either CNS WHO grade 2 or grade 3 to an ependymoma, according to its histopathological features. In an integrated diagnosis, CNS WHO grade can be presented in a specific tier (eg, Tables 4–6).22
Choroid Plexus Tumors
The classification of choroid plexus tumors remains largely unchanged, although this family of tumors has been separated from the category of primary neuroepithelial tumors that feature more glial and/or neuronal differentiation and less epithelial differentiation.
Medulloblastomas
WHO CNS5 has altered the classification of medulloblastomas to mirror new knowledge of their clinical and biological heterogeneity. Initially, consensus established 4 principal molecular groups: WNT-activated, sonic hedgehog (SHH)-activated, group 3, and group 4.42 WNT and SHH medulloblastomas were included in the 2016 classification, and SHH tumors divided on the basis of TP53 status (with TP53-mutant and TP53-wildtype tumors having markedly different clinicopathological characteristics). Non-WNT/non-SHH medulloblastomas comprised group 3 and group 4 tumors. These groups are represented in WHO CNS5 (Table 1); however, through large-scale methylation and transcriptome profiling, new subgroups have emerged at a more granular level below the 4 principal molecular groups: 4 subgroups of SHH and 8 subgroups of non-WNT/non-SHH medulloblastomas.43–47 Like the 4 principal molecular groups of medulloblastoma, some of these subgroups are associated with clinicopathological and genetic features that provide clinical utility, having either diagnostic, prognostic, or predictive value. One example is the delineation of 2 (out of 4) SHH subgroups, SHH-1 and SHH-2, both dominated by medulloblastomas from young children.47,48 These subgroups show significantly different outcomes, and recent clinical trial data suggest that specific chemotherapeutic regimens can help those patients with tumors in the poor prognosis subgroup49,50 indicating that these distinctions may be predictive rather than solely prognostic.
The histopathological classification of medulloblastoma listed in the 2016 WHO classification comprised 4 morphologic types: classic, desmoplastic/nodular, medulloblastoma with extensive nodularity (MBEN), and large cell/anaplastic. These have now been combined into 1 section that describes them as morphologic patterns of an inclusive tumor type, Medulloblastoma, histologically defined (Table 1). The morphologic differences have their own specific clinical associations,51–54 and molecularly defined medulloblastomas demonstrate distinct associations with the morphologic patterns. For example, all true desmoplastic/nodular medulloblastomas and MBENs align with the SHH molecular group,55 and most are in the SHH-1 and SHH-2 subgroups.47 Nearly all WNT tumors have classic morphology, and most large cell/anaplastic tumors belong either to the SHH-3 subgroup or to the Grp3/4 subgroup 2.46
Given their heterogeneity and the need to classify medulloblastomas according to a combination of histopathological and molecular features, these tumors should be reported in a layered and integrated format. NOS and NEC options also exist for these lesions in the appropriate settings.
Other Embryonal Tumors
The other embryonal tumors (ie, aside from Medulloblastoma) are AT/RT; Embryonal tumor with multilayered rosettes (ETMR); CNS neuroblastoma, FOXR2-activated; and CNS tumor with BCOR internal tandem duplication (ITD; Figure 5). Whereas AT/RT and ETMR were included in previous WHO classifications, CNS neuroblastoma, FOXR2-activated and CNS tumor with BCORITD are new to CNS5. In addition, CNS5 recognizes 3 molecular subtypes of AT/RT and an ETMR with DICER1 alteration (in addition to the more common C19MC type). CNS tumors with BCOR ITD are now included in WHO CNS5 as embryonal tumors, but these neoplasms are not definitively neuroectodermal. Exon 15 BCOR ITDs have been reported in several morphologically similar sarcomas, and there is currently no consensus as to whether these tumors should be considered neuroepithelial or mesenchymal neoplasms; the nosology of such tumor types may need to change in light of future findings. CRINET has been introduced as a provisional entity within this category, and the broad designation CNS embryonal tumor is included for embryonal tumors that defy a more specific diagnosis, ie, that are NEC or NOS.12,13 Given the histological and molecular complexity sometimes found in these lesions, an integrated, tiered diagnostic report is helpful for transparent and effective communication of relevant tumor characteristics.22,31
Pineal Tumors
Pineal gland tumors are neoplasms that include Pineocytoma, Pineal parenchymal tumor of intermediate differentiation (PPTID), and Pineoblastoma, as well as Papillary tumor of the pineal region (PTPR). An addition to WHO CNS5 is Desmoplastic myxoid tumor of the pineal region, SMARCB1-mutant (Figure 6), a rare SMARCB1-mutant tumor lacking histopathological signs of malignancy.56 Many questions remain about the behavior of pineal tumors, and histological grading criteria for PPTID, PTPR, and Desmoplastic myxoid tumor, SMARCB1-mutant are yet to be defined.
Importantly, molecular studies play a role in their diagnoses. For example, KBTBD4 in-frame insertions are a desirable criterion for the diagnosis of PPTID.57 Using methylation profiling, pineoblastomas can be divided into 4 molecular subtypes: Pineoblastoma, miRNA processing-altered 1 in children and characterized by DICER1, DROSHA, or DGCR8 mutations; Pineoblastoma, miRNA processing-altered_2 mostly in older children with a relatively good prognosis and also featuring DICER1, DROSHA, or DGCR8 mutations; Pineoblastoma, MYC/FOXR2-activated, in infants and having MYC activation and FOXR2 overexpression; and Pineoblastoma, RB1-altered, in infants and with similarities to retinoblastoma.58,59
Meningiomas
Meningioma is considered a single type in WHO CNS5, with its broad morphological spectrum reflected in 15 subtypes. It is now emphasized that the criteria defining atypical or anaplastic (ie, grade 2 and 3) meningioma should be applied regardless of the underlying subtype. As in prior classifications, chordoid and clear cell meningioma are noted to have a higher likelihood of recurrence than the average CNS WHO grade 1 meningioma and have hence been assigned to CNS WHO grade 2; however, larger and prospective studies would be helpful to validate these suggested CNS WHO grade 2 assignments and to suggest additional prognostic biomarkers. In addition, historically, rhabdoid and papillary morphology qualified for CNS WHO grade 3 irrespective of any other indications for malignancy. While papillary and rhabdoid features are often seen in combination with other aggressive features, more recent studies suggest that the grading of these tumors should not be on the basis of a rhabdoid cytology or papillary architecture alone.60 Several molecular biomarkers are also associated with classification and grading of meningiomas, including SMARCE1 (clear cell subtype), BAP1 (rhabdoid and papillary subtypes), and KLF4/TRAF7 (secretory subtype) mutations, TERT promoter mutation61 and/or homozygous deletion of CDKN2A/B62 (CNS WHO grade 3), H3K27me3 loss of nuclear expression63 (potentially worse prognosis), and methylome profiling64 (prognostic subtyping).
Mesenchymal, Non-Meningothelial Tumors
WHO CNS5 has strived to align the terminology of mesenchymal, non-meningothelial tumors with their counterparts in the WHO Blue Book on Bone and Soft Tissue Tumors. WHO CNS5 also now only covers those entities that occur uniquely in the CNS or, though similar to their soft tissue counterparts, are encountered regularly in the CNS. Some common soft tissue tumors that can exceptionally be found in the CNS (eg, leiomyoma) are no longer included given that their diagnostic features are identical to their soft tissue counterparts. New types that have been added are Intracranial mesenchymal tumor, FET-CREB fusion-positive (provisional); CIC-rearranged sarcoma; and Primary intracranial sarcoma, DICER1-mutant (Figure 7). The term “hemangiopericytoma” has been retired, with the tumor now termed only Solitary fibrous tumor (rather than the hybrid term “Solitary fibrous tumor/hemangiopericytoma” used in the 2016 CNS classification). This term now aligns with the soft tissue nomenclature, although the newly modified 3-tiered CNS grading scheme remains a site-associated difference.
Nerve Tumors
Several changes were made to the classification of nerve tumors. Because paragangliomas involve specialized neuroendocrine cells of the sympathetic and parasympathetic nervous system, these tumors are now included with nerve tumors. Also, given both immunohistochemical and DNA methylation differences and the lack of familial associations, the paraganglioma of the cauda equina/filum terminale region is now recognized as a distinct tumor type from the more common paragangliomas encountered in other sites. Furthermore, it is now appreciated that the previously designated “melanotic schwannoma” is a highly distinctive and frequently aggressive tumor type with unique genetic underpinnings that distinguish it from all other nerve sheath tumors, including schwannomas; in accordance with the soft tissue classification, its name has been changed to Malignant melanotic nerve sheath tumor. Lastly, a new subtype has been added to the neurofibroma section: Atypical neurofibromatous neoplasm of unknown biological potential (ANNUBP) is an NF1-associated tumor with worrisome features of malignant transformation that is still quantitatively insufficient for a definitive diagnosis of Malignant peripheral nerve sheath tumor (MPNST).
Lymphomas and Histiocytic Tumors
WHO CNS5 only includes those lymphoid and histiocytic tumor entities that occur relatively often in the CNS or that have special histological or molecular features when they occur in the CNS. The complete spectrum of these neoplasms is covered in the corresponding Blue Book on Classification of Tumors of Haematopoietic and Lymphoid Tissues.
Tumors of the Sellar Region
In past editions, Adamantinomatous craniopharyngioma and Papillary craniopharyngioma were considered subtypes (variants) of craniopharyngioma, whereas they are now classified as distinct tumor types, given their different clinical demographics, radiologic features, histopathologic findings, genetic alterations, and methylation profiles.65,66 On the other hand, Pituicytoma, Granular cell tumor, and Spindle cell oncocytoma are included in 1 section as a related group of tumor types67; although they may represent morphologic variations of the same tumor, patient demographics, and clinical outcomes vary and so they are still classified separately.68
For pituitary adenomas, WHO CNS5 follows the guidelines of the fourth-edition endocrine WHO classification, dividing these tumors by their adenohypophyseal cell-lineage according to combined immunohistochemical expression of pituitary hormones and transcription factors. WHO CNS5 also includes the new term Pituitary neuroendocrine tumor (PitNET) proposed by the WHO endocrine group, which will be further debated for the fifth-edition WHO classification of endocrine tumors.69 Lastly, Pituitary blastoma (Figure 8), a rare embryonal neoplasm of infancy composed of primitive blastemal cells, neuroendocrine cells, and Rathke epithelium, has been added as a tumor type in WHO CNS5.
Metastatic Tumors
The section on metastatic tumors is divided into those that preferentially affect the brain and spinal cord parenchyma vs those that favor the meninges. Given progress in the treatment of specific systemic cancers, attention has been paid to those immunohistochemical and molecular diagnostic markers that are helpful for diagnosis and/or for guiding therapies of these tumors.
Genetic Tumor Syndromes
Although genetic tumor syndromes are not part of the official WHO CNS5 classification (eg, they are not in Table 1), those tumor syndromes that characteristically feature nervous system neoplasms are included in the fifth-edition CNS Blue Book. This section has been expanded, now covering 8 disorders not covered in the prior Blue Book.
Conclusions
All classifications are imperfect representations, reflecting the state of understanding in a field at a particular time as well as the interpretations of that information by limited numbers of experts. WHO CNS5, like its predecessors, should therefore be seen as a work in progress, as a stage in the evolution of CNS tumor classification. WHO CNS5 has attempted to introduce new knowledge into the classification in as careful but progressive a manner as possible, by including newly recognized entities, by phasing out ostensibly obsolete tumor types, and by adjusting the taxonomic structure. It is hoped that such changes and their explanations provide practical guidance to pathologists and specialists in neuro-oncology around the world and that such progress benefits the patients who are affected by CNS tumors.
Funding
None.
Conflict of interest statement. The authors have no conflicts of interest relating to the information presented in the manuscript. The content of this article represents the personal views of the authors and does not represent the views of the authors’ employers and associated institutions. Where authors are identified as personnel of the International Agency for Research on Cancer/World Health Organization, the authors alone are responsible for the views expressed in this article and they do not necessarily represent the decisions, policy or views of the International Agency for Research on Cancer/World Health Organization.
Authorship statement. All authors contributed to generating the materials expressed in this review, which are part of the 2021 WHO Classification of CNS Tumors. All authors participated in the writing of the article as well as the generation of tables and figures.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}